


Infantile Spasms
John F. Kerrigan, MD
Division of Neurology, Barrow Neurological Institute, St. Joseph’s Hospital and Medical Center, Phoenix, Arizona
Abstract
Infantile spasms, a peculiar form of epilepsy affecting young children, usually begin between 3 and 24 months of age. They consist of clusters of body jerks and typically occur on awakening. Most children with infantile spasms are retarded, sometimes severely, depending on the underlying cause. The pathogenesis is unknown but probably reflects a complex interaction between cortical and subcortical structures. Despite being associated with significant potential side effects, adrenocorticotropic hormone (ACTH) remains the drug of first choice in the United States. In contrast, vigabatrin has emerged as the drug of choice in most other developed countries. Many patients with infantile spasms have focal pathology and may be candidates for epilepsy surgery, and their developmental profile may improve after successful epilepsy surgery.
Key Words: adrenocorticotropic hormone, epilepsy, infantile spasms, vigabatrin
The first recorded reference to infantile spasms is particularly memorable and poignant. In 1841, Dr. W. J. West published a letter in Lancet regarding “a very rare and singular species of convulsion peculiar to young children.”52 West accurately and vividly described infantile spasms and the developmental and cognitive decline often associated with them. He stated: “As the only case I have witnessed is in my own child, I shall be very grateful to any member of the profession who can give me any information on the subject, either privately or through your excellent Publication.” Unfortunately, progress with respect to understanding and treating this disorder has been modest, and the desperation of parents of affected children remains just as palpable today.
Clinical Features
 Infantile spasms are distinguished by the appearance of the seizure itself and the age group in which they typically occur. The spasms consist of sudden, shock-like contractions, involving widespread muscle groups of the trunk, neck, and extremities. Typically, there is a flexion or sudden “drop” to the torso or head while both arms are simultaneously uplifted. Continuous video-electroencephalographic (video-EEG) monitoring has characterized individual spasms showing their variability between (and within) individuals and defining flexor, extensor, and mixed flexor-extensor groups.32,33 The spasm can persist in a brief tonic phase, typically lasting 2 seconds but sometimes as long as 10 seconds. Although it is important to be aware that individual spasms can vary, these differences seem to have little predictive value with respect to etiology or course. Asymmetrical or unilateral spasms can occur and suggest the possibility of focal cerebral pathology, which in turn should suggest the possibility of surgical treatment (see below).
Infantile spasms are distinguished by the appearance of the seizure itself and the age group in which they typically occur. The spasms consist of sudden, shock-like contractions, involving widespread muscle groups of the trunk, neck, and extremities. Typically, there is a flexion or sudden “drop” to the torso or head while both arms are simultaneously uplifted. Continuous video-electroencephalographic (video-EEG) monitoring has characterized individual spasms showing their variability between (and within) individuals and defining flexor, extensor, and mixed flexor-extensor groups.32,33 The spasm can persist in a brief tonic phase, typically lasting 2 seconds but sometimes as long as 10 seconds. Although it is important to be aware that individual spasms can vary, these differences seem to have little predictive value with respect to etiology or course. Asymmetrical or unilateral spasms can occur and suggest the possibility of focal cerebral pathology, which in turn should suggest the possibility of surgical treatment (see below).
Spasms occur in a group or cluster as much as 80% of the time.32 The tendency of spasms to cluster represents one of the most important historical features in making the diagnosis. Individual spasms are observed 5 to 30 seconds apart, and the child often relaxes or cries during these intervals. Typically, the number of spasms in an individual cluster varies between 5 and 20. Occasionally, however, more than 100 can be observed. The intensity of the spasms is also highly variable, sometimes increasing, then decreasing, during a single cluster. Some spasms are subtle—little more than a momentary arrest of activity or a slight nod of the head. Clusters of spasms occur most often when the child awakens from sleep.
Most children with infantile spasms become symptomatic between 3 and 8 months old38 and rarely after 12 months. The spasms usually resolve by the age of 2 to 3 years but occasionally persist into late childhood.49 Although the spasms resolve, at least 50% to 60% of patients will continue to have epilepsy and to develop other types of seizures. Approximately half of these patients will develop the atypical absence seizures, tonic seizures, and slow spike-wave EEG pattern that characterize the Lennox-Gastaut syndrome.31
Etiology
Infantile spasms is an epilepsy syndrome of early childhood with multiple and widely divergent causes. It is customary to classify these causes as idiopathic, cryptogenic, and symptomatic. The list of diseases that can be associated with infantile spasms in the symptomatic group is extensive (Table 1) and reinforces the concept that a final, common, “downstream” pathophysiological mechanism peculiar to this age group must cause the epileptic spasms. Diffuse or multifocal disorders (such as hypoxic injury) and focal disorders (such as tumor, stroke, or abscess) can both cause typical infantile spasms in this age group.
The idiopathic subgroup appears to occur in otherwise normal infants, presumably with a hereditary predisposition to infantile spasms.51 A family history for other forms of idiopathic epilepsy or febrile convulsions often exists. The development of these infants and their head growth at the time of spasm onset are normal. They respond favorably to treatment, or the spasms resolve spontaneously. Long-term follow-up suggests a normal developmental outlook for this group, which may comprise about 10% of the population with infantile spasms. The gene or genes involved with this syndrome have not been identified.
Almost without exception, the spasms of the cryptogenic and symptomatic groups are more refractory to treatment. Ultimately, at least 90% of affected infants will be mentally retarded. The cryptogenic group includes infants with an underlying brain disease but its exact etiology defies identification. The symptomatic group includes those in whom the exact cause has been identified. The incidence of symptomatic spasms has increased at the expense of cryptogenic spasms,50 particularly since the advent of magnetic resonance (MR) imaging, which can identify cerebral pathology that might have been missed with computed tomography.
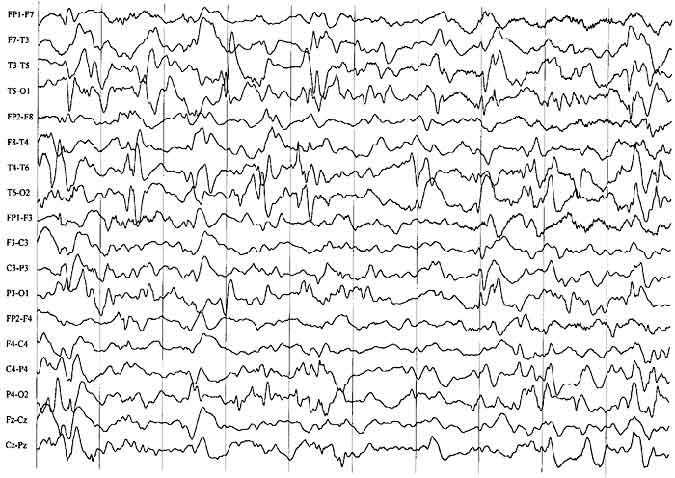
EEG Findings
The interictal EEG associated with infantile spasms is usually characterized by a distinctive pattern called hypsarrhythmia23 or by one of its variant forms.27 This pattern consists of a very high-amplitude, chaotic, poorly organized background with large numbers of sharp and spike transients (Fig. 1). These spikes occur independently over many areas, most predominately over the posterior region of the head. During non-rapid eye movement (non-REM) sleep, the tracing tends to become more fragmentary and discontinuous. However, the EEG may appear normal or almost normal during REM sleep, a finding not yet fully explained. Not all patients with spasms show hypsarrhythmia. Perhaps a third show other patterns, such as focal or lateralized slowing or spiking. This type of finding should prompt close review of the MR imaging study to search for a potentially resectable lesion.
An ictal EEG is recorded when a cluster of spasms is captured during the tracing. A number of different patterns may be seen.32 The most common, however, is a generalized electrodecremental pattern, consisting of sudden flattening of the EEG from its high-amplitude hypsarrhythmic baseline. This electrodecrement can be proceeded by a high-amplitude generalized slow wave. As with the interictal EEG, the ictal EEG should be examined carefully in a search for possible focal features.
Pathophysiology
The pathophysiology for infantile spasms is unknown, and satisfactory animal models have not yet been developed. Any comprehensive pathophysiological model for infantile spasms must explain the following observations. First, the syndrome is usually very tightly linked to a particular period of development (“age-specific”). Second, infantile spasms can be caused by a wide array of underlying causes, including focal, diffuse, or multifocal pathologies (“etiology nonspecific”). Finally, a model must account for the peculiar response of this particular epilepsy syndrome to adrenocorticotropic hormone (ACTH) or glucocorticoid treatment and its general resistance to the usual anticonvulsant medications.
Recent hypotheses have emphasized neuroanatomical relationships, highlighting the concept that the production of infantile spasms most likely requires interaction between cortical and subcortical structures. Using flourodeoxyglucose positron emission tomography (FDG-PET), Chugani and colleagues discovered bilateral activation of the lenticular nuclei (and possibly brainstem structures) in a large percentage of infants with spasms, irrespective of etiology.11 Based on characteristic sleep disturbances in children with infantile spasms, Hrachovy and coworkers had earlier speculated about the importance of pontine influences.26 Thus far, neurochemical studies to elucidate a possible abnormality in neurotransmitter systems have been unrewarding.42
Recently, a neuroendocrine hypothesis for the origin of infantile spasms has been proposed.4 Corticotropin-releasing factor (CRF) is elaborated by the hypothalamus as a nonspecific response to stress, as might occur with a variety of cerebral insults. CRF is known to be a potent proconvulsant agent in animal models, particularly in immature animals.6 CRF receptors are particularly abundant during the first postnatal week in the developing rat brain.30 ACTH or glucocorticoid treatment may decrease the release of CRF from the hypothalamus by feedback inhibition. Conceptually, this hypothesis should be testable with a therapeutic trial with CRF antagonist agents.
Treatment
Infantile spasms are particularly resistant to the standard anticonvulsant drugs. Exogenous administration of ACTH was discovered to be an effective therapy in 195847 and was the treatment of choice until recently.7 Nonetheless, the dosing regimen and duration of therapy have been controversial.
The high-dose regimen was popularized by Snead and colleagues,46 who treated spasms with a dose of ACTH, standardized for body surface area, at 150 units/m2/day, with a 90% response rate. Subsequently, Hrachovy and coworkers25 randomized subjects to either a high-dose or a low-dose regimen of 20 to 30 units/day and evaluated the response with intermittent video-EEG monitoring. They found no difference between the two treatment groups with a 50% and 58% response rate, respectively. This same group previously reported a double-blind, placebo-controlled, crossover study that compared ACTH (low-dose, at 20 to 30 units/day) with oral prednisone (2 mg/kg/day).28 No significant differences with respect to stopping spasms were observed.
Bringing these conflicting data full circle, Baram and colleagues recently randomized patients to receive either high-dose ACTH (150 units/m2/day) or prednisone (2 mg/kg/day) and assessed them with intermittent video-EEG analysis.5 A positive response consisted of both cessation of spasms and elimination of hypsarrhythmia on video-EEG monitoring. Of 15 infants treated with ACTH, 13 (87%) responded while only 4 of 14 infants (29%) in the prednisone group had positive outcomes.
The potential for side effects with ACTH is significant, particularly with a high-dose regimen.45 Difficulties can include the development of a cushingoid phenotype, weight gain with fluid retention, hypertension, and glucose intolerance. Less frequently, the risk of developing an infection (thrush is particularly common), gastritis, or hypertrophic cardiomyopathy can increase. The risk of adrenal suppression increases with the duration of treatment. These potential complications must be actively monitored during follow-up.34
My routine for the initial treatment of infantile spasms is the same, regardless of whether the patient appears to be in the symptomatic, cryptogenic, or idiopathic subgroup. For the first three or four days, patients receive ACTH in a low or conventional dose of 40 units/day (approximately 75 units/m2/day). If the patient fails to respond (spasms do not cease), the dose is increased to the high-dose regimen of 150 units/m2/day, usually for two weeks, and then tapered over 2 to 3 weeks. If a relapse occurs, a second course of ACTH is given, again with the high-dose regimen, but it is tapered at a slower rate.
The effectiveness of other medications for treating infantile spasms, including valproic acid,3,44 felbamate,29 nitrazepam (an investigational drug in the United States),9,16 and pyridoxine,40,48 has also been studied. Other therapies such as intravenous administration of immunoglobulins18 and the ketogenic diet37 have also been reported. Despite being associated with fewer adverse events, these therapies have failed to demonstrate the effectiveness of ACTH treatment in small (and usually uncontrolled) trials.
As an alternative therapy to ACTH for children with infantile spasms, vigabatrin has generated some legitimate excitement. It is emerging as the drug of first choice, supplanting ACTH in countries where it is available. Vigabatrin acts by irreversibly binding to and inhibiting gamma-aminobutyric acid transaminase (GABA-T), the major degradative enzyme for GABA, thereby increasing brain concentrations of this inhibitory neurotransmitter.
Chiron and colleagues treated 70 children with refractory infantile spasms (age range, 2 months to 13 years at treatment) with vigabatrin.10 With a mean follow-up of slightly longer than 3 months, 43% of the patients were free of seizures and seizure frequency had improved at least 50% in 68% of the patients. A subsequent uncontrolled, retrospective study examined the European experience with vigabatrin as the initial therapy for infantile spasms1 and found that 68% of the infants responded with at least initial complete suppression of spasms. At the end of the study (median, 7.6 months), 50% of the infants remained free of seizures. Based upon this experience and the recognition that vigabatrin is typically well tolerated in children,21 Aicardi et al. have suggested that vigabatrin should be used as the initial therapy of choice for patients with newly diagnosed infantile spasms.1
Vigabatrin is still an investigational drug in the United States. It is available in both Mexico and Canada. Recent findings of a possible retinopathy associated with the use of vigabatrin19 have raised new concerns about its safety. Consequently, ACTH remains the drug of first choice for infantile spasms in the United States.
Surgical Treatment
Recognition of the role of focal cerebral pathology in the genesis of infantile spasms is increasing. Although infantile spasms are usually conceptualized as a generalized epilepsy, several studies combining clinical and EEG features have highlighted that many children also have focal features. The spasms may be asymmetrical,43 or partial seizures can precede or co-exist with infantile spasms41 or even occur simultaneously.15 EEG recordings can show focal or asymmetrical features either ictally or interictally.14,17,22
Structural imaging studies, particularly MR imaging, can show focal or regional pathology in patients with infantile spasms.50 A large variety of focal lesions has been identified, although focal encephalomalacia and focal cortical dysplasia are the most common (Table 1). Chugani and coworkers have identified a subgroup of patients with focal or regional abnormalities on FDG-PET but with no abnormalities on MR imaging who have proved to have cortical dysplasia by pathological examination after epilepsy surgery.13 That FDG-PET is more sensitive than MR imaging in detecting potential lesions associated with focal cortical dysplasia has been confirmed.20,36 Together, these clinical, electrographic, and imaging indicators of focal disease have occurred in as many as 66% of patients with infantile spasms at one referral center.36 A much smaller number, however, prove to be suitable candidates for epilepsy surgery.
Since the initial case reports that described successful surgery for infantile spasms,8 several series have been reported. Palm and colleagues reported 10 infants with infantile spasms or Lennox-Gastaut syndrome and porencephalic cysts who improved after the cyst cavities were unroofed surgically.39
Chugani and co-workers have reported their surgical experience with 23 infants at the University of California in Los Angeles (UCLA).12 At surgery, 17 patients had active spasms, and all were evaluated with continuous video-EEG monitoring, MR imaging, and FDG-PET. In 14 patients, an abnormality was identified only by PET. In the other patients, abnormalities were also detected by MR imaging. Intraoperative electrocorticography was used to help determine the surgical margin in patients scheduled for focal cortical resection (it was not used in the hemispherectomy cases). Seizures were not monitored with chronic intracranial electrodes. At a mean final follow-up of 28 months, 65% of the patients were free of seizures. In 17% of the patients, seizure control improved at least 75%. Four patients (17%) failed to benefit from the procedure.
Wyllie reported 12 infants with catastrophic localization-related epilepsies (some of whom had infantile spasms).53 In this series, 75% were either free of seizures or had rare seizure events after resective surgery.
In summary, the clinical assessment for newly diagnosed patients with infantile spasms should include a careful review of their history, EEG patterns, and MR imaging studies for indicators that might suggest a focal origin. In patients with cryptogenic spasms who fail to respond to initial medical management, FDG-PET is indicated to look for focal cortical dysplasia, a potentially resectable lesion. The actual preoperative evaluation (and, ultimately, the surgical procedure itself) is highly tailored to the individual circumstances and pathology and is best managed by referral centers experienced with this process.
Outcome
Unfortunately, the outcome for many patients with infantile spasms remains poor. Mental retardation is observed in 90% of the cases and may be severe or very severe in two-thirds of patients.24 The 10% of patients who are ultimately determined to have normal long-term outcomes probably belong to the idiopathic group as discussed above. The factors that determine long-term developmental outcome are controversial. By consensus, the underlying etiology is the single most important determinant. Most studies do not demonstrate a significant difference with regard to timing or type of treatment,24 but some reports have suggested that developmental outcome is worse if initial treatment is delayed.35
The UCLA group has recently reported their 2-year follow-up of developmental outcomes in children undergoing surgery for infantile spasms.2 Developmental levels, as determined with the Vineland Adaptive Behavior Scale, improved significantly 2 years after surgery compared to preoperative levels, and they compared favorably with historical controls. The course of individual patients in the series, however, varied considerably.
References
- Aicardi J, Mumford JP, Dumas C, et al: Vigabatrin as initial therapy for infantile spasms: A European retrospective survey. Sabril IS Investigator and Peer Review Groups. Epilepsia 37:638-642, 1996
- Asarnow RF, LoPresti C, Guthrie D, et al: Developmental outcomes in children receiving resection surgery for medically intractable infantile spasms. Dev Med Child Neurol 39:430-440, 1997
- Bachman DS: Use of valproic acid in treatment of infantile spasms. Arch Neurol 39:49-52, 1982
- Baram TZ: Pathophysiology of massive infantile spasms: Perspective on the putative role of the brain adrenal axis. Ann Neurol 33:231-236, 1993
- Baram TZ, Mitchell WG, Tournay A, et al: High-dose corticotropin (ACTH) versus prednisone for infantile spasms: A prospective, randomized, blinded study. Pediatrics 97:375-379, 1996
- Baram TZ, Schultz L: Corticotropin-releasing hormone is a rapid and potent convulsant in the infant rat. Brain Res Dev Brain Res 61:97-101, 1991
- Bobele GB, Bodensteiner JB: The treatment of infantile spasms by child neurologists. J Child Neurol 9:432-435, 1994
- Branch CE, Dyken PR: Choroid plexus papilloma and infantile spasms. Ann Neurol 5:302-304, 1979
- Chamberlain MC: Nitrazepam for refractory infantile spasms and the Lennox-Gastaut syndrome. J Child Neurol 11:31-34, 1996
- Chiron C, Dulac O, Beaumont D, et al: Therapeutic trial of vigabatrin in refractory infantile spasms. J Child Neurol Suppl 2:S52-S59, 1991
- Chugani HT, Shewmon DA, Sankar R, et al: Infantile spasms: II. Lenticular nuclei and brain stem activation on positron emission tomography. Ann Neurol 31:212-219, 1992
- Chugani HT, Shewmon DA, Shields WD, et al: Surgery for intractable infantile spasms: Neuroimaging perspectives. Epilepsia 34:764-771, 1993
- Chugani HT, Shields WD, Shewmon DA, et al: Infantile spasms: I. PET identifies focal cortical dysgenesis in cryptogenic cases for surgical treatment. Ann Neurol 27:406-413, 1990
- Donat JF, Lo WD: Asymmetric hypsarrhythmia and infantile spasms in West syndrome. J Child Neurol 9:290-296, 1994
- Donat JF, Wright FS: Simultaneous infantile spasms and partial seizures. J Child Neurol 6:246-250, 1991
- Dreifuss F, Farwell J, Holmes G, et al: Infantile spasms. Comparative trial of nitrazepam and corticotropin. Arch Neurol 43:1107-1110, 1986
- Drury I, Beydoun A, Garofalo EA, et al: Asymmetric hypsarrhythmia: Clinical electroencephalographic and radiological findings. Epilepsia 36:41-47, 1995
- Echenne B, Dulac O, Parayre-Chanez MJ, et al: Treatment of infantile spasms with intravenous gamma-globulins. Brain Dev 13:313-319, 1991
- Eke T, Talbot JF, Lawden MC: Severe persistent visual field constriction associated with vigabatrin. BMJ 314:180-181, 1997
- Ferrie CD, Maisey M, Cox T, et al: Focal abnormalities detected by 18FDG PET in epileptic encephalopathies. Arch Dis Child 75:102-107, 1996
- Fisher RS, Kerrigan JF: Vigabatrin: Toxicity, in Levy RH, Mattson RH, Meldrum BS (eds): Antiepileptic Drugs, Fourth Edition. New York: Raven, 1995
- Gaily EK, Shewmon DA, Chugani HT, et al: Asymmetric and asynchronous infantile spasms. Epilepsia 36:873-882, 1995
- Gibbs FA, Gibbs EL: Atlas of Electroencephalography: Epilepsy. Cambridge, MA: Addison-Wesley, 1952
- Glaze DG, Hrachovy RA, Frost JD, Jr., et al: Prospective study of outcome of infants with infantile spasms treated during controlled studies of ACTH and prednisone. J Pediatr 112:389-396, 1988
- Hrachovy RA, Frost JD, Jr., Glaze DG: High-dose, long-duration versus low-dose, short-duration corticotropin therapy for infantile spasms. J Pediatr 124:803-806, 1994
- Hrachovy RA, Frost JD, Jr., Kellaway P: Sleep characteristics in infantile spasms. Neurology 31:688-693, 1981
- Hrachovy RA, Frost JD, Jr., Kellaway P: Hypsarrhythmia: Variations on the theme. Epilepsia 25:317-325, 1984
- Hrachovy RA, Frost JD, Jr., Kellaway P, et al: Double-blind study of ACTH vs prednisone therapy in infantile spasms. J Pediatr 103:641-645, 1983
- Hurst DL, Rolan TD: The use of felbamate to treat infantile spasms. J Child Neurol 10:134-136, 1995
- Insel TR, Battaglia G, Fairbanks DW, et al: The ontogeny of brain receptors for corticotropin-releasing factor and the development of their functional association with adenylate cyclase. J Neurosci 8:4151-4158, 1988
- Jeavons PM, Harper JR, Bower BD: Long-term prognosis in infantile spasms: A follow-up report on 112 cases. Dev Med Child Neurol 12:413-421, 1970
- Kellaway P, Hrachovy RA, Frost JD, Jr., et al: Precise characterization and quantification of infantile spasms. Ann Neurol 6:214-218, 1979
- King DW, Dyken PR, Spinks IL, Jr., et al: Infantile spasms: Ictal phenomena. Pediatr Neurol 1:213-218, 1985
- Kongelbeck SR: Discharge planning for the child with infantile spasms. J Neurosci Nurs 22:238-244, 1990
- Koo B, Hwang PA, Logan WJ: Infantile spasms: Outcome and prognostic factors of cryptogenic and symptomatic groups. Neurology 43:2322-2327, 1993
- Kramer U, Sue WC, Mikati MA: Focal features in West syndrome indicating candidacy for surgery. Pediatr Neurol 16:213-217, 1997
- Livingston S, Eisner V, Pauli L: Minor motor epilepsy: Diagnosis, treatment and prognosis. Pediatrics 21:916-928, 1958
- Lombroso CT: A prospective study of infantile spasms: Clinical and therapeutic correlations. Epilepsia 24:135-158, 1983
- Palm DG, Brandt M, Korinthenberg R: West syndrome and Lennox-Gastaut syndrome in children with porencephalic cysts: Long-term follow-up after neurosurgical treatment, in Niedermeyer E, Degen R (eds): The Lennox-Gastaut Syndrome. New York: Alan R. Liss, 1988, pp 419-426
- Pietz J, Benninger C, Schafer H, et al: Treatment of infantile spasms with high-dosage vitamin B6. Epilepsia 34:757-763, 1993
- Plouin P, Dulac O, Jalin C, et al: Twenty-four-hour ambulatory EEG monitoring in infantile spasms. Epilepsia 34:686-691, 1993
- Pranzatelli MR: Putative neurotransmitter abnormalities in infantile spasms: Cerebrospinal fluid neurochemistry and drug effects. J Child Neurol 9:119-129, 1994
- Shewmon DA: Ictal aspects with emphasis on unusual variants, in Dulac O, Chugani HT, Dalla Bernardina B (eds): Infantile Spasms and West Syndrome. London: W.B. Saunders, 1994, pp 36-51
- Siemes H, Spohr HL, Michael T, et al: Therapy of infantile spasms with valproate: Results of a prospective study. Epilepsia 29:553-560, 1988
- Snead OC: Other antiepileptic drugs: Adrenocorticotrophic hormone (ACTH), in Levy RH, Mattson RH, Meldrum BS (eds): Antiepileptic Drugs, Fourth Edition. New York: Raven, 1995
- Snead OC, 3rd, Benton JW, Jr., Hosey LC, et al: Treatment of infantile spasms with high-dose ACTH: Efficacy and plasma levels of ACTH and cortisol. Neurology 39:1027-1031, 1989
- Sorel L, Dusaucy-Bauloye A: A propos de 21 cas d’hypsarythmia de Gibbs. Son traitement spectaculaire par l’ACTH. Acta Neurol Psychiatr Belg 58:130-141, 1958
- Takuma Y, Seki T: Combination therapy of infantile spasms with high-dose pyridoxal phosphate and low-dose corticotropin. J Child Neurol 11:35-40, 1996
- Talwar D, Baldwin MA, Hutzler R, et al: Epileptic spasms in older children: Persistence beyond infancy. Epilepsia 36:151-155, 1995
- van Bogaert P, Chiron C, Adamsbaum C, et al: Value of magnetic resonance imaging in West syndrome of unknown etiology. Epilepsia 34:701-706, 1993
- Vigevano F, Fusco L, Cusmai R, et al: The idiopathic form of West syndrome. Epilepsia 34:743-746, 1993
- West WJ: On a particular form of infantile convulsions. Lancet 1:724-725, 1841
- Wyllie E, Comair YG, Kotagal P, et al: Epilepsy surgery in infants. Epilepsia 37:625-637, 1996