
Issues Related to the Study of Malignant Brain Tumors
Joan Rankin Shapiro, PhD
Division of Neurology, Barrow Neurological Institute, St. Joseph’s Hospital and Medical Center, Phoenix, Arizona
Abstract
Cancer appears to result from the accumulation of multiple genetic alterations. Malignant brain tumors likewise develop sequential aberrations that contribute to malignant progression. Brain cancer is unregulated cell proliferation with the additional property of invasiveness. Consequently, a broader spectrum of characterizations with the biology of brain tumors must be considered to determine which of the dynamic mechanisms creates the malignant phenotype. This article reviews the different aspects of brain tumor biology and the techniques developed to assess these changes.
Key Words : apoptosis, cell cycle, genetic code, genetic instability, gliomas, oncogenes, tumor heterogeneity, tumor suppressor genes
Cancer is a multistep process in which a normal cell becomes transformed, immortalized, and invasive. Once immortalized, tumor cells appear to have the ability to proliferate without the normal constraints governing cell division. The genetic aberrations associated with this process are best known for leukemias19 and lymphomas18 Similar information about solid tumors has lagged behind. Some of the difficulty in detecting genetic changes in solid tumors (i.e., malignant brain tumors) relates to tissue availability, histological grading, tumor heterogeneity, and the ability to obtain direct preparations from informative samples. Despite these difficulties, our understanding is being advanced by new technologies that require less tissue for analysis and that permit the use of archival samples, which often supply extensive history about patients.
Figure 1. Representative karyotype from a freshly resected glioblastoma multiforme tumor. This karyotype illustrates both the numerical and structural abnormalities that can exist in a single tumor cell. The karyotype deviation is as follows: 68,XX,-1,der(3)t(3;7)(p21;q12)x2, +4,+4,+4,+5,+5,+5,+6,del(6)(q13),+7,+7,+7,+7,+7,del(7)(q11.1)x2,+9,-10,+11,+12,+12,+14,-17,der(18)(15;18)(q15;p11.3)x2,+19,+20,+21,+21.
The Genetic Code
All characteristics (i.e., hair color, height, intelligence, susceptibility to disease) are determined by the genetic code, a language that communicates how proteins are built. In humans the genetic code resides within 46 chromosomes (22 pairs of autosomes and a pair of sex chromosomes) located in the nucleus of every nucleated cell. Chromosomes are visible under light microscopy only when a cell prepares to divide; otherwise, they remain as elongated strands. By definition, a chromosome is a single DNA molecule with its associated histone and nonhistone proteins.
A DNA molecule communicates information through a linear sequence of nitrogenous bases that compose genes that encode the information needed to build a specific protein. In humans, the genetic code is comprised of only four nitrogenous bases: adenine (A), thymine (T), cytosine (C), and guanine (G). The size of the smallest gene is about 2000 nitrogenous bases, and the average gene is about 20,000 to 30,000 nitrogenous bases long. The 46 DNA molecules contain approximately 100,000 genes, each of which codes for a protein that will either become part of a cell’s structure (structural protein), regulate another gene (regulatory genes), or serve a catalytic function (enzymes).
Some genes have alternative forms called alleles. Each gene or allele occupies a specific position or location on a DNA molecule (chromosome) known as the genetic locus. DNA molecules exist as pairs (one DNA molecule donated by the mother, the other by the father); therefore, two genes are responsible for coding each protein.
Genes underlie an individual’s genotype or genetic composition; the phenotype is the observed result of the expression of a particular gene or set of genes. Currently, about 5,000 phenotypes have been identified in humans and are presumed to be inherited in a Mendelian fashion.
Genetic aberrations or mutations can occur to any gene. Many mutations reflect an abnormality of a single nitrogenous base pair (point mutation) within a gene. Mutations can also involve a segment of the DNA molecule and hence more than one gene (i.e., translocations, deletions, inversions or duplications). A specific type of a genetic aberration and its location underlie each of the 3,500 known human genetic diseases.38 A small fraction of these genetic abnormalities can be carried in the germ line (hereditable mutations). Most diseases, however, are caused by sporadic DNA mutations that accumulate throughout an individual’s lifetime. Therefore, any cell in the human body capable of cell division has the potential of undergoing genetic alteration(s) that can transform it from a normal cell into a cancer cell.
Methods for Analyzing Genetic Material
Many investigative approaches can be used to define the genetic alterations that cause a cancer.
Cytogenetic Analysis
One of the first characterizations of malignant brain tumors was cytogenetic analysis.63 It consists of collecting chromosomes from dividing cells; banding the chromosomes with specific enzymes, dyes, or both; and arranging them into a karyotype (Fig. 1). The karyotype is like a “fingerprint” of the genomic content of a tumor cell. The chromosome number and rearrangements of chromosomes in the karyotype (i.e., translocations, deletions, inversions, and duplications) are important elements in this analysis. Chromosome preparations from freshly resected tissue represent a particular time point in the malignant progression of a tumor cell.
When multiple patients with a particular diagnosis are studied, specific recurring abnormalities can be determined. Such abnormalities provide clues about the location of genes instrumental in producing the cancer phenotype. Cytogenetic analysis is also helpful in deciphering the progressive changes associated with malignant gliomas, but the analysis depends upon a cell entering cell division. Even the most malignant tumor rarely has more than 2 to 5% of the cells undergoing cell division at any one time. Therefore, one must assume that most of the cells have a similar genetic make-up.
Molecular Cytogenetics
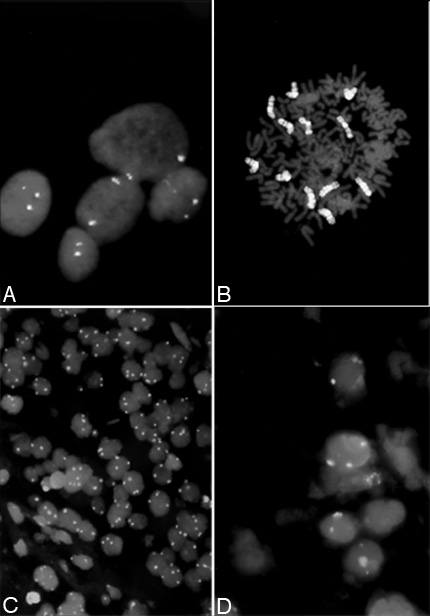
Molecular cytogenetics, a new technology that uses fluorescent probes to analyze chromosomal abnormalities, is rapidly advancing our ability to determine more about a tumor population. The technique is called fluorescent in situ hybridization (FISH) and has the advantage of detecting numerical and structural aberrations in both nondividing and dividing tumor cells. This method permits most of the cells in a sample to be evaluated, thereby reducing the need for large amounts of tissue. FISH uses a chromosome-specific DNA fluorescent probe to distinguish one chromosome from another (Fig. 2). The fluorescent probe can be designed to detect the centromere of a specific chromosome (Fig. 2A) or the entire chromosome (chromosome painting) (Fig. 2B). Furthermore, the study can be completed in 10 to 12 hours while standard cytogenetics requires 2 or more weeks.
Comparative Genomic Hybridization
Comparative genomic hybridization (CGH) also uses fluorescent probes to detect gains and losses of chromosomes. DNA samples obtained from a tumor and normal control cells (typically lymphocytes or skin fibroblasts from the same patient) are labeled with a different fluorescent reporter molecule such as fluorescein isothiocyanate (FITC) or tetra-rhodamine isothiocyanate (TRITC). With the reporter molecule attached to the DNA, equal amounts of the tumor and control DNA are mixed and hybridized to normal metaphase chromosome preparations together with an excess of unlabeled human DNA. The unlabeled DNA competes for the highly repetitive sequences in the human genome by binding to these common sequences. This step is important because investigators wish to analyze only the unique sequences of a specimen.
The principle of CGH reflects that in normal cells each genetic locus has two genes that code for a specific protein. However, tumor DNA may have multiple copies of a single gene or lose one or both copies of a gene. CGH detects this gain or loss in gene copy number if it occurs in the majority of cells. After hybridization, the ratio of the fluorescence intensities is measured along individual chromosomes and quantitated by computer-assisted digital image analysis to provide an indication of the relative changes in copy number (number of genes).
Figure 3. Hypothetical cell retaining different fragments from chromosome 7. Each fragment would be retained as part of a structurally rearranged chromosome. The overlapping regions of these fragments will appear as an amplification on the line tracing illustrated to the left of the chromosome 7 idiogram. CGH = comparative genomic hybridization.
Because CGH is a study of tumor cell populations, it cannot provide information about an individual cell or segment in a single cell’s chromosome as can a karyotype prepared by standard cytogenetic analysis. However, CGH can identify the map position or region of chromosomal amplification and/or loss that occurs in the majority of cells. Consider a hypothetical cell retaining a number of fragments from chromosome 7 (Fig. 3). Each fragment can occur as part of a derivative chromosome (rearranged chromosome), a marker chromosome (a chromosome that cannot be identified by conventional banding techniques), or both. This hypothetical cell with the chromosome 7 fragments is representative of the majority of cells in the tumor population being analyzed. The overlapping regions containing the same genes represent genetic material that is being amplified (gained) in the cancer cell. The retention of these extra fragments translates into amplification of a region(s) or a gene(s) that CGH can detect. This technique has been used to analyze the genetic changes in a number of tumor systems, including gliomas.15,54
Spectral Karyotyping
Spectral karyotyping uses different dye combinations that enable each human chromosome to be colored differently.55 Thus, all 22 chromosome pairs, in addition to the X and Y chromosomes, can be distinguished from each other by using an interferometer to measure their emitted light spectra. Computer software converts this information into a visual image in which each human chromosome is assigned a different color.
The advantage of spectral karyotyping is the ease with which abnormalities of chromosome structure can be detected. In very malignant tissue, standard banding and FISH technologies often fail to identify the chromosomes involved in the complex structural rearrangements. Because each chromosome is a different color, even very small segments of chromosomes involved in complex rearrangements and/or in marker chromosomes can easily be identified.
Techniques from Molecular Biology
Standard molecular biological techniques include methods that detect abnormalities of gene copy number or gene expression in addition to abnormalities of the protein encoded by these genes. Southern blot hybridization detects the presence and number of genes in a population of cells. By measuring the levels of mRNA, Northern blot hybridization determines if genes are being expressed. Western blot hybridization determines if genes have been translated into cellular protein. Each technique requires a probe that detects the specific gene, its mRNA, or the protein it produces. The probe can be radioactive, a fluorescent compound, or an antibody, or it can involve an enzymatic staining reaction.
Irrespective of the probe used, each technique is limited by its level of sensitivity. Therefore, each assay depends upon the level of expression of the gene, mRNA, and/or protein, all of which must be produced in great enough quantities to be detected. Newer techniques like the polymerase chain reaction (PCR) make it possible to detect a single copy of a gene by amplifying the gene a million or more times in less than several hours. These PCR-amplified products can be subjected to restriction fragment length polymorphism (RFLP), microsatellite instability, or single-strand conformation polymorphism (SSCP) analyses that permit the investigator to determine if a gene has been mutated, lost, or both. Other techniques permit cloning and characterization of genes before their function has been defined: PCR-based differential mRNA display, subtractive hybridization or denaturation-renaturation techniques, and cDNA expression array.
Although this battery of techniques can be used to detect aberrant gene function in tumors, it is also crucial to determine if only a small population of cells generates the aberrant expression or whether it is a common feature of the cells. Consequently, immunohistochemistry is used to detect protein products in cells, and in situ hybridization is used to detect mRNA from fresh, frozen, or paraffin-embedded material. Data generated in the test tube can then be used to confirm the pattern of expression in the tissue.
Tumor Heterogeneity
To be successful, tissue analysis must account for tumor heterogeneity and the proportion of normal cells in the sample. Fresh, frozen, or paraffin-embedded brain tumor tissue contains normal reactive astrocytes, endothelial cells, smooth muscle cells, and meningeal cells in addition to the tumor cells. The proportion of normal cells to tumor cells becomes an issue when only small populations of tumor cells carry the genetic abnormalities. Since each technique is limited by its sensitivity, small populations of cells with a specific abnormality might fall below the threshold of detection of a particular technology. Therefore, tissue samples must be assessed histologically to determine the proportion of normal cells to abnormal cells. Otherwise, a negative result might be mistakenly attributed to the absence of a phenotype rather than to the limited sensitivity of a given test.
Tumor cell heterogeneity refers to cellular variation in the observed genotype and/or phenotype of a tumor population and can be either cellular or regional. Cells immediately adjacent to one another may have a very different histological appearance. These cellular differences are generated because the parental population is genetically unstable and subject to mechanisms that generate aneuploidy,69 endopolyploidization, or both.65
Regional differences develop when tumor cells begin to invade the surrounding normal brain. During migration, some tumor cells will develop additional abnormalities that confer a selective advantage for growth. Proliferation ensues. As this process is repeated, a tumor is seeded with microfoci that are both genotypically and phenotypically different.11
Pathologists first documented tumor heterogeneity as a fundamental feature of gliomas,46,53 and this finding has been reflected in cytogenetic analyses,58,60 flow cytometry DNA analyses,11 oncogene expression,20 growth factors and receptor gene copy number and expression,52,56 proliferation markers,10 various glioma-associated antigens,3 and intrinsic resistance to chemo- and radiotherapy.1,59,61 Thus, all investigations of a gene or gene product must consider these problems.10,11
Regional heterogeneity also has a significant impact on pathological diagnosis. The analysis of small, nonrepresentative tumor biopsies often results in misclassifications,13,21 and 20 to 25% of the cases are misdiagnosed as a lower grade.46 In general, heterogeneity increases with tumor grade, making it difficult to develop universally applicable histopathological criteria.
Phenotypes differ not only from one tumor to another but also within a tumor. Furthermore, the expression of any given phenotype can be affected by multiple factors. For example, markers such as Ki-67 or MIB-1 demonstrate that proliferative activity varies considerably.10 The observed differences in the staining pattern of these markers could reflect clonal cellular differences or tumor cells interacting with the local hypoxic microenvironment. The latter develops because the vasculature cannot keep pace with a growing neoplasm.51 Likewise, the overexpression of one phenotype does not reflect any other phenotype. Thus, the staining pattern of a proliferative marker such as Ki-67 or MIB-1 may not reflect amplification of epidermal growth factor receptor (EGFR).
Patterns of heterogeneity, however, have suggested how gliomas evolve in a tumor mass.11 A regional study of a low-grade oligoastrocytoma based on both cytogenetic and flow cytometry analyses found that populations of cells in adjacent regions were similar. Further from these regions, new clonal karyotypic deviations were observed or additional abnormalities were acquired in a clonal population. These findings are indicative of expansive infiltration, presumably by a more aggressive clone(s), and support that tumors progress by multiple local mutations and clonal expansion.45
The mechanisms underlying this genetic variability are areas of intense research. Normal cells are expected to have a background mutation rate of 1.4 x 10-10. In tumor cells, however, the mutation rate far exceeds this figure.47 Explanations have been sought for this disparity. DNA is replicated with exceptional accuracy, which is safeguarded by a number of sequential mechanisms. A breakdown in any one of these mechanisms has been attributed to genomic instability.
Figure 4. Representation showing how the accumulation of genetic aberrations causes cellular and regional tumor heterogeneity in brain tumors. Note that some tumor populations as well as normal cells are lost as progression proceeds. The original mutation in this example (outlined) is lost as a single mutation by the final phase of progression shown here. The process, however, continues, and the original mutation is still present with secondary and tertiary mutations.
Genetic Instability
Genomic instability results from mutation or exogenous interference with the metabolic pathways that govern the replication (duplication) and division of DNA. Genomic instability is best described for DNA regions called microsatellites—short, repeated nucleotide sequences interspersed throughout the entire genome. This repeating unit can be as short as two nucleotides, although trinucleotides (CAG or CTG) are more common. In normal individuals, the repeating unit may be 15 to 20. In diseased states it can be twice that amount or more. Analysis of microsatellites has helped to determine gene abnormalities involved in cancer tissue. Thirteen different neurodegenerative abnormalities, such as Huntington’s disease and Machado-Joseph disease, have been identified by this type of analysis.43,67
Tandem repeats can involve larger segments of DNA and have been labeled minisatellites.6 These tandem repeats are unstable because they are prone to slippage during DNA replication. A small loop forms in either the template or nascent DNA strand, and this phenomenon initiates the loss of genetic material that contributes to the breakdown of pathways guarding the fidelity of replication. It also generates the environment necessary for additional mutations, karyotypic alterations, or recombinations to occur.
In brain tumors, genomic instability appears to be more important to progression rather than to the initiation of a tumor.12 The concept that tumors progress to a more malignant state by the acquisition of mutations is not new (Fig. 4). Nowell45 suggested this hypothesis in 1976. Only within the last 10 years, however, has the acquisition of specific genetic alterations that lead to clonal expansion and selection by local environmental factors been demonstrated. Since each analysis performed on a tissue sample reflects a single time point in that tumor’s evolution, many tumors must be analyzed to determine the proper sequence of these genetic aberrations.
Other mechanisms that might create genomic instability have been explored. Hastie and coworkers23 suggested that these changes related to chromosome telomeres. Telomeres are repeated sequences that ‘cap’ the ends of every chromosome. In humans, the repeat unit forming this cap is TTAGGG. As a cell prepares to divide, DNA polymerases replicate each linear strand of DNA producing sister chromatids that divide. However, DNA polymerases cannot replicate DNA all the way to the tip of the DNA strand—a small piece (the end of the telomere) is left uncopied. If chromosomes had no mechanism to compensate for this feature, a chromosome would shorten with each cell division. Eventually, the elimination of genes would prove lethal to a cell.
The enzyme, telomerase, adds a piece of DNA to the 3´ end of DNA.4 This addition protects a chromosome from being shortened during the replication process. In many cancers, telomerase is overexpressed. Some investigators have therefore suggested that it plays a critical role in maintaining the immortalization of cancer cells.29 Aneuploidy, produced by the abnormal segregation of sister chromatids, is also a form of genetic instability. A recent report has demonstrated that mutations in the telomerase template block chromosome separation during anaphase.30 This mechanism could generate the numerous segregational errors observed in cancer and in malignant gliomas.57 In gliomas, telomerase is seldom amplified although about 60% of gliomas show alterations in telomeric length.5 A much more common event observed in the cytogenetic analysis of gliomas involves telomeric fusion (i.e., the ends of chromosomes stick together). The meaning of this abnormality is not fully understood,14 but it could serve as a mechanism by which genes are lost.
In humans, many genes are responsible for repairing replication errors. A breakdown in this pathway could also generate genomic instability. A defect in the mismatch repair genes is thought to be the direct cause of hereditary nonpolyposis colorectal cancer.35 One defective allele is inherited through the germ line, and the second is obtained as a somatic mutation. With the advent of the second mutation, the control of replicative accuracy declines and mutations accumulate in genes at a much higher frequency than normal.
Loeb has hypothesized that cancer is manifested by a mutator phenotype34 that requires a tumor cell to acquire the requisite number of changes in DNA to progress through the multistep carcinogenic process. Thus, an adenoma with a mutator phenotype will proceed more quickly to a carcinoma because of an increased frequency of mutations that can affect oncogenes and tumor suppressor genes critical to the malignant progression of this cancer. In gliomas, each chromosome containing the map location of the mismatch repair genes is involved in chromosome rearrangement, loss, or both (Table 1).57 DNA repair genes32 associated with chemoresistance are described in Genetics of Primary Malignant Gliomas in this issue.
Oncogenes and Tumor Suppressor Genes
Two general categories of genes are directly involved in transformation and progression of the malignant process by one or more of the mechanisms described above. Oncogenes are genes capable of transforming a eukaryotic cell into a cell that grows in a manner analogous to tumor cells, while tumor suppressor genes are genes capable of suppressing tumor formation. Both groups of genes play important roles in the development of the nervous system7 and in response to DNA damage.41
Humans possess DNA sequences (genes) similar to viral oncogenes (genes capable of transforming normals cells). When functioning normally, these genes are called proto-oncogenes. Proto-oncogenes code for proteins that are involved in a signal transduction pathway (a pathway designed to carry messages from the cell’s surface to the nucleus where it initiates an effect) or that serve as a growth factor (proteins that stimulate a cell to divide) or a growth factor receptor (cell surface proteins that transmit messages through the cell membrane).
Proto-oncogenes become oncogenes when they are activated by mutation or by increasing the copy number (gene amplification). These genes act in a dominant manner—only one allele at that genetic locus needs to be altered for this aberrant expression to occur. Irrespective of the mechanism that triggers a proto-oncogene to become an oncogene, the result is an increase in the protein product capable of affecting the growth-stimulatory pathway or apoptosis. Approximately 100 oncogenes have been identified, several of which are aberrantly expressed in brain tumors (Table 2).
Tumor suppressor genes function exactly the opposite of oncogenes. Their function is to slow growth or to suppress it entirely. Several mechanisms underlie the failure of a tumor suppressor gene to function normally. First, a mutation can inactivate the function of the gene. Second, the gene or a whole chromosome can be lost through genetic recombination or the aberrant segregation of chromosomes. Both genes must be lost and/or inactivated before the abnormal phenotype, which removes the ability of these protein products to regulate proliferation, is expressed. Eliminating these control mechanisms is an essential step in tumor formation. Several genes in the tumor suppressor gene family are known to be aberrant in human gliomas; other genetic loci that contain putative tumor suppressor genes await identification (Table 3).
The first evidence that loss of a tumor suppressor gene causes tumor formation came from retinoblastomas, neoplasms that can occur in either familial or sporadic forms. In the familial form, an individual inherits one mutated gene; mutation to the second allele occurs at a later point as a somatic mutation. The inactivation or loss of both alleles causes tumor formation.31 With the sporadic form of retinoblastoma, both mutations must occur at the somatic level. Consequently, this form of retinoblastoma typically appears in older children. In the 1970s, Knudson developed the two-hit hypothesis as a means of explaining the genetic events required to cause a retinoblastoma to develop.31 Almost a decade later, molecular techniques became available that allowed Cavenee and coworkers to show that both alleles were lost or inactivated in retinoblastoma (Rb gene) patients and that one gene was inherited through the germ line in the familial form.8
The Cell Cycle and Apoptosis
A major question to be addressed is how oncogenes, growth factors, and tumor suppressor genes contribute to neoplastic disease. The multistep process of transforming a normal cell, either in vitro37 or in vivo,68 requires a series of changes in both oncogenes and tumor suppressor genes. For many years research concentrated on the notion that an oncogene or tumor suppressor gene could initiate transformation as a single entity. Both groups of genes, however, interact with the growth control pathway(s) and elicit their effect through its breakdown.
Figure 5. Diagram illustrating the steps in the cell cycle. These steps are composed of an orderly sequence of phases regulated by a whole series of different genes. Cells destined to proliferate enter the G1 phase of the cell cycle, which is considered the first checkpoint. Passing through this phase, the cell enters the S phase in which the DNA is replicated. The G2 phase assesses for errors in the replication process before the cell is allowed to begin the mitotic phase. The mitotic step consists of prophase (chromosome coiling), metaphase (chromosomes are organized onto the mitotic spindle), anaphase (division of the sister chromatids) and telophase (completes the division of the parental cell into two daughter cells). The specific cyclins associated with each phase are indicated on the periphery.
The Cell Cycle
Mitotic figures are one of the hallmarks of malignancy in brain tumors, especially in high-grade tumors. However, the signals that initiate the commitment of a tumor cell to divide occurs well before mitotic figures appear in the cell cycle (Fig. 5). Each phase of the cell cycle is controlled by several families of proteins that activate or inhibit the progression of events in the pathway. One group of highly conserved proteins, called cyclins, binds to cyclin-dependent kinases (CDKs) to form protein kinases. The cyclin family of proteins consists of cyclin A and B, D1-3 and E. Cyclin levels fluctuate with specific phases of the cell cycle, and they determine the timing of the activation of CDKs and their substrate specificity (Fig. 5).
Activation of the cyclin/CDKs heterodimer complex requires a series of phosphorylations and dephosphorylations to specific amino acid residues by cyclin-dependent activating kinases (CAKs) and phosphatases, two additional groups of proteins in this pathway. Phosphorylation or dephosphorylation alters the conformation of the cyclin/CDK molecule and modifies the kinase activity.40 For example, the cyclin B1/CDK1 complex phosphorylates the H1 histone. The activation of this protein kinase mediates the condensation of chromosomes in the prophase stage of the mitotic cycle.44
The family of CDK inhibitors (CDKIs) modulates the activity of the cyclins and CDKs by suppressing their function. Consequently, these inhibitors are considered candidates for tumor suppressor genes. Two major groups of CDKIs exist—the p21-like family (p21, p27, and p57) and the p16-like family (p16, p15, p18 and p19). The two groups are defined by their homology, CDK targets, and mechanism of inhibition.24
The cell cycle is an orderly sequence of phases (G0®G1®S®G2®M), which have checkpoints to determine if the cell should proceed to the next step (Fig. 5). G0 cells, such as neurons, undergo terminal differentiation soon after birth and never re-enter the cell cycle. Upon receiving the appropriate signal, quiescent cells that have not undergone terminal differentiation enter the cell cycle. The first phase, G1, is characterized by cell growth and the synthesis of cellular proteins necessary for DNA synthesis. G1 is usually the longest phase of the cell cycle. During the S phase each DNA molecule is replicated or synthesized. Each strand of DNA becomes two strands (sister strands) joined at a single region known as the centromere or kinetochore on a chromosome. This region is the last region to replicate just before the two strands separate during anaphase. Briefer than either the G1 or S phase is the G2 phase, the phase that determines if DNA replication is complete. Once replication is completed, the cell enters the mitotic phase, which consists of prophase (chromosome coiling), metaphase (assembling the chromosomes on the middle of the mitotic spindle), anaphase (the separation of the sister chromatids to opposite poles), and telophase (the division of the cytoplasm into two cells). Normal glia cells, which divide to permit the growth, repair, and replacement of cells, complete the cell cycle in about 24 hours.49 In contrast, some brain tumor cells complete the cycle in 12 hours or less.33
Apoptosis
Another pathway is designed to destroy normal and abnormal cells. Programmed cell death (apoptosis) is a genetically controlled cascade of cellular events that leads to the elimination of cells. When more cells are made than can be used or when cells are damaged by ionizing radiation, ultraviolet irradiation, cytotoxic drugs, mutagens, or carcinogens, the damage is either repaired at one of the checkpoints in the cell cycle or the cells are directed to the apoptotic pathway for elimination.
The proteins that control the apoptotic pathway either suppress cell death or induce it. The bcl proteins promote cell survival. The two key proteins in this family, bcl-2 and bcl-X, suppress apoptosis by blocking the release of cytochrome c from mitochondria. If cytochrome c is released, the ICE (interleukin 1b-converting enzyme) family of proteins (recently renamed caspases 1-10), which cause cytoplasmic and chromatin condensation and DNA fragmentation, are activated. Both bad and bax are proteins that promote cell death by blocking the function of the bcl-2 family of genes. Phosphorylation and dephosphorylation of each of these proteins are also critical events in the activation or inhibition of this pathway.
Apoptosis is an important mechanism in animal development and in diseases such as cancer. When the genes controlling apoptosis are deregulated, cells develop inappropriate survival and tumorigenic properties. The TP53 gene located on chromosome 17 (17p13.1) is a key gene in this decision-making process of whether cells survive or die.25,39 This gene codes for the nuclear protein p53, which regulates whether cells survive or die after their DNA is damaged by radio- or chemotherapy. The p53 protein is involved in at least two checkpoints in the cell cycle, the G1®S and the G2®M transitions (Fig. 5). These checkpoints are stages at which DNA damage can be repaired before a cell proceeds to the next phase.
Which pathway is selected for a cell appears to be related to the cell type and the DNA damage incurred. Therefore, p53 is important in the treatment of cancer patients. Chemotherapy drugs, such as 1, 3-bis-(2-chlorethyl)-1-nitrosourea (BCNU) and cisplatin, induce DNA damage that can activate the p53 apoptotic pathway and cause the transformed cells to be eliminated. For such treatments to be effective, normal p53 protein must be present in the cell. Unfortunately, the gene coding for this protein is often mutated in human cancer,22 including brain cancer.26
Abnormal p53 proteins can be generated from a variety of mechanisms.9 Individuals can inherit an abnormal TP53 gene through the germ line, develop a somatic mutation, undergo a deletion of the chromosome 17p, or develop a structural chromosome rearrangement or a genomic insertion. Each of these events can cause the loss of p53 protein or a mutated form. Normal p53 protein can also be inactivated if it forms a protein complex with viral oncoproteins or a cellular gene product [e.g., murine double minute 2 (MDM2) protein]. In a tumor cell, MDM2 can be amplified, overexpressed, or both. This protein will bind to wild-type p53.50 The binding of MDM2 to p53 inactivates the wild-type p53 so that it cannot interact with other proteins.
More than a third to half of all grade II to IV malignant gliomas have a mutation in TP53 or have lost one or both alleles.16,36 About 10% of tumors with normal p53 have an amplification of MDM2 that suppresses the normal function of TP53 gene.2 In adult26 and pediatric gliomas,48,64 abnormal TP53 is one of the most common aberrations. An early change in the evolution of gliomas,26 this aberration is typically associated with a less favorable prognosis.22
The gene TP53 can independently promote cell survival or cell death. It can also act in concert with other gene products to accomplish this task. Some genes upregulate TP53 (e.g., c-myc), others inactivate it, and still others respond to its up- or downregulation. The upregulation of p53 causes the downregulation of insulin-like growth factor-I (IGF-I). Like other growth factors, IGF-I is considered a survival factor necessary for cell growth or proliferation. A reduction in survival factor(s) such as IGF-I may limit the signals that promote a cell’s survival and thereby induce the apoptotic pathway. However, knowledge of how p53 coordinates the interaction between apoptosis (cell death) and proliferation (cell survival) is still preliminary.
For many years, p53 was thought to have no close relatives. The discovery of p73 protein, which appears to share the same growth-inhibiting and apoptosis-promoting effects identified with p53, has changed that conception.27,28 The TP73 gene encodes two isoforms, p73a and p73b, that differ at their C-termini because of alternative mRNA splicing. The homology between these genes is 63% in the DNA-binding region, suggesting that p73 might also be a sequence-specific transactivator that recognizes DNA targets much like p53. Many questions about the tumor suppressive nature of p53 remain. Its chromosomal location on 1p36 is a region often deleted in malignant gliomas, further suggesting its potential importance in the evolution of gliomas.
Apoptotic pathways independent of p53 also exist. Members of the tumor necrosis factor (TNF) family of genes, such as fas and TNF-related apoptosis-inducing ligand (TRAIL/Apo2L), induce apoptosis in many cancer cells lines. Like p53-dependent pathways, fas and TRAIL/Apo2L induce apoptosis through the activation of caspases42,62 that mediate DNA fragmentation.
Thus, research continues to define the genetic lesions associated with uncontrolled proliferation. Understanding how and where gene(s) function can provide the means to eradicate tumor cells in the future. Once an aberrant gene is identified, the next step is to determine which protein it encodes and how the protein functions in a cell. The identity of a specific nonrandom genetic change in a tumor system can become a diagnostic-prognostic marker, and each genetic change has the potential of becoming a target for the design of new therapies. Furthermore, it is now possible to associate these genetic changes with specific tumor grades, suggesting that some of these genetic aberrations are critical events in tumor progression17,66 as discussed in the following article, Genetics of Primary Malignant Gliomas.
References
- Benediktsson G, Blomquist E, Carlsson J: Heterogeneity in cell loss and frequency of slow growing colonies of human glioma cell lines. Some effects of radiation. Anticancer Res 9:1483-1488, 1989
- Biernat W, Kleihues P, Yonekawa Y, et al: Amplification and overexpression of MDM2 in primary (de novo) glioblastomas. J Neuropathol Exp Neurol 56:180-185, 1997
- Bigner DD, Bigner SH, Pontén J, et al: Heterogeneity of genotypic and phenotypic characteristics of fifteen permanent cell lines derived from human gliomas. J Neuropathol Exp Neurol 40:201-229, 1981
- Blackburn EH, Greider CW: Telomeres. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 1995
- Bondy M, Wiencke J, Wrensch M, et al: Genetics of primary brain tumors: A review. J Neurooncol 18:69-81, 1994
- Buard J, Jefferys AJ: Big, bad minisatellites. Nature Genet 15:327-328, 1997
- Burek MJ, Oppenheim RW: Programmed cell death in the developing nervous system. Brain Pathology 6:427-446, 1996
- Cavenee WK, Dryja TP, Phillips RA: Expression of recessive alleles by chromosomal mechanisms in retinoblastomas. Nature 305:779-784, 1983
- Chang F, Syrjanen S, Syrjanen K: Implications of the p53 tumor-suppressor gene in clinical oncology. J Clin Oncology 13:1009-1022, 1995
- Coons SW, Johnson PC: Regional heterogeneity in the proliferative activity of human gliomas as measured by the Ki-67 labeling index. J Neuropathol Exp Neurol 42:609-618, 1993
- Coons SW, Johnson PC, Shapiro JR: Cytogenetic and flow cytometry DNA analysis of regional heterogeneity in a low grade glioma. Cancer Res 55:1569-1577, 1995
- Dams E, Van de Kelft EJZ, Martin JJ, et al: Instability of microsatellites in human gliomas. Cancer Res 55:1547-1549, 1995
- Daumas-Duport C: Histological grading of gliomas. Current Opinion Neurol Neurosurg 5:924-931, 1992
- de Lange T: Telomere dynamics and genome instability in human cancer, in Blackburn EH, Greider CW (eds): Telomeres. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 1995, pp 265-293
- Feuerstein BG, Mohapatra G: Molecular cytogenetic quantitation of gains and losses of genetic material from human gliomas. J Neurooncol 24:47-55, 1995
- Frankel RH, Bayona W, Koslow M, et al: p53 mutations in human malignant gliomas: Comparison of loss of heterozygosity with mutation frequency. Cancer Res 52:1427-1433, 1992
- Furnari FB, Huang HJS, Cavenee WK: Genetics and malignant progression of human brain tumors, in Ponder BAJ, Cavenee WK, Solomon E (eds): Genetics and Cancer: A Second Look. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 1995, pp 233-275
- Gaidano G, Dalla-Favera R: Molecular biology of lymphoid neoplasms, in Mendelsohn J, Howley PM, Israel MA, et al. (eds): The Molecular Basis of Cancer. Philadelphia: W.B. Saunders, 1995, pp 251-293
- Gauwerky CE, Croce CM: Molecular genetics and cytogenetics of hematopoietic malignancies, in Mendelsohn J, Howley PM, Israel MA, et al. (eds): The Molecular Basis of Cancer. Philadelphia: W.B. Saunders, 1995, pp 18-31
- Gerosa MA, Talarico D, Fognani C, et al: Overexpression of N-ras oncogene and epidermal growth factor receptor gene in human glioblastomas. J Natl Cancer Inst 81:63-67, 1989
- Glantz MJ, Burger PC, Herndon JE, et al: Influence of the type of surgery on the histologic diagnosis in patients with anaplastic gliomas. Neurology 41:1741-1744, 1991
- Harris CC, Hollstein M: Clinical implications of the p53 tumor-suppressor gene. N Engl J Med 329:1318-1327, 1993
- Hastie ND, Dempster M, Dunlop MG, et al: Telomere reduction in human colorectal carcinoma and with ageing. Nature 346:866-868, 1990
- Hunter T, Pines J: Cyclins and cancer: II-cyclin D and CDK inhibitors come of age. Cell 79:573-582, 1994
- Isobe M, Emanuel BS, Givol D, et al: Localization of gene for human p53 to band 17p13. Nature 320:84-85, 1986
- James CD, Carlbom E, Dumanski JP, et al: Clonal genomic alterations in glioma malignancy stages. Cancer Res 48:5546-5551, 1988
- Jost CA, Marin MC, Kaelin WG, Jr: p73 is a human p53-related protein that can induce apoptosis. Nature 389:191-194, 199
- Kaghad M, Bonnet H, Yang A, et al: Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 90:809-819, 1997
- Kim NW, Piatyszek MA, Prowse KR, et al: Specific association of human telomerase activity with immortal cells and cancer.Science 266:2011-2015, 1994
- Kirk KE, Harmon BP, Reichardt IK, et al: Block in anaphase chromosome separation caused by a telomerase template mutation.Science 275:1478-1481, 1997
- Knudson AG Jr: Mutation and cancer: Statistical study of retinoblastomas. Proc Natl Acad Sci 68:820-823, 1971
- Liang BC, Ross DA, Reed E: Genomic copy number changes of DNA repair genes ERCC1 and ERCC2 in human gliomas. J Neurooncol 26:17-23, 1995
- Lindgren A, Westermark B, Pontén J: Serum stimulation of stationary human glia and glioma cells in culture. Exp Cell Res 95:311-319, 1975
- Loeb L: Microsatellite instability: Marker of a mutator phenotype in cancer. Cancer Res 54:5059-5063, 1994
- Lothe RA, Peltomaki P, Meling GI, et al: Genomic instability in colorectal cancer: Relationship to clinicopathological variables and family history. Cancer Res 53:5849-5852, 1993
- Louis DN: The p53 gene and protein in human brain tumors. J Neuropathol Exp Neurol 53:11-21, 1994
- McCormick JJ, Maher VM: Malignant transformation of mammalian cells in culture, including human cells. Environ Mol Mutagen 14 Suppl 16:105-113, 1989
- McKusick VA: Mendelian Inheritance in Man. Baltimore, MD: John Hopkins University Press, 1994
- Miller C, Mohandas T, Wolf D, et al: Human p53 localized to short arm of chromosome 17. Nature 319:783-784, 1986
- Morgan DO: Principles of CDK regulation. Nature 374:131-134, 1995
- Morgan SE, Kastan MB: p53 and ATM: Cell cycle, cell death, and cancer. Adv Cancer Res 71: 1-25, 1997
- Nagata S, Golstein P: The Fas death factor. Science 267:1449-1456, 1995
- Néri C, Albanèse V, Lebre A-S, et al: Survey of CAG/CTG repeats in human cDNAs representing new genes: Candidates for inherited neurological disorders. Human Mol Genet 5:1001-1009, 1996
- Nigg EA: Targets of cyclin-dependent kinases. Curr Opin Cell Biol 5:187-193, 1993
- Nowell PC: The clonal evolution of tumor cell populations. Science 194:23-28, 1976
- Paulus W, Peiffer J: Intratumoral histologic heterogeneity of gliomas. A quantitative study. Cancer 64:442-447, 1989
- Perucho M: Microsatellite instability: The mutator that mutates the other mutator. Nature Med 2:630-631, 1996
- Pollack IF, Hamilton RL, Finkelstein SD, et al: The relationship between TP53 mutations and overexpression of p53 and prognosis in malignant gliomas of childhood. Cancer Res 57:304-309, 1997
- Pontén J, Westermark B: Properties of human malignant glioma cells in vitro. Med Biol 56:184-193, 1978
- Reifenberger G, Liu L, Ichimura K, et al: Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res 53:2736-2739, 1993
- Reynolds TY, Rockwell S, Glazer PM: Genetic instability induced by the tumor microenvironment. Cancer Res 56:5754-5757, 1996
- Scheck AC, Mehta BM, Beikman MK, et al: BCNU-resistant human glioma cells with over-representation of chromosomes 7 and 22 demonstrate increased copy number and expression of platelet-derived growth factor genes. Genes Chrom Cancer 8:137-148, 1993
- Scherer HJ: The forms of growth in gliomas and their practical significance. Brain 63:1-35, 1940
- Schrock E, Thiel G, Lozanova T, et al: Comparative genomic hybridization of human malignant gliomas reveals multiple amplification sites and nonrandom chromsomal gains and losses. Am J Pathol 144:1203-1218, 1994
- Schröck E, du Manoir S, Veldman T, et al: Multicolor spectral karyotyping of human chromosomes. Science 273:494-497, 1996
- Shapiro JR, Mehta BM, Ebrahim SAD, et al: Tumor heterogeneity and intrinsically chemoresistant subpopulations in freshly resected human malignant gliomas, in Sudilovsky O, Pitot HC, Liotta LA (eds): Boundaries Between Promotion and Progression During Carcinogenesis. New York: Plenum Press, 1991, pp 243-262
- Shapiro JR, Scheck AC: Brain tumors, in Wolman SR, Sell S (eds): Cytogenetic Markers of Human Cancer. New Jersey: Humana Press, Inc., 1997, pp 319-368
- Shapiro JR, Shapiro WR: The subpopulations and isolated cell types of freshly resected high grade human gliomas: Their influence on the tumor’s evolution in vivo and behavior and therapy in vitro. Cancer Metast Rev 4:107-124, 1985
- Shapiro JR, Shapiro WR, Mohamed AN, et al: BCNU-sensitivity in parental cells and clones from four freshly resected near-diploid human gliomas: An astrocytoma, an anaplastic astrocytoma and two glioblastomas multiforme. J Neurooncol 15:209-227, 1993
- Shapiro JR, Yung W-KA, Shapiro WR: Isolation, karyotype, and clonal growth of heterogeneous subpopulations of human malignant gliomas. Cancer Res 41:2349-2359, 1981
- Shapiro WR, Yung WA, Basler GA, et al: Heterogeneous response to chemotherapy of human gliomas grown in nude mice and as clones in vitro. Cancer Treat Rep 65:55-59, 1981
- Sheridan JP, Marsters SA, Pitti RM, et al: Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors.Science 277:818-821, 1997
- Spriggs AI, Boddington MM, Clarke CM: Chromosomes of human cancer cells. Br Med J 1431-1435, 1962
- Sure U, Rüedi D, Tachibana O, et al: Determination of p53 mutations, EGFR overexpression, and loss of p16 expression in pediatric glioblastomas. J Neuropathol Exp Neurol 56:782-789, 1997
- Therman E, Sarto GE, Stubblefield PA: Endomitosis: A reappraisal. Am J Hum Genet 63:13-18, 1983
- von Deimling A, Louis DN, Wiestler OD: Molecular pathways in the formation of gliomas. Glia 15:328-338, 1995
- Warren ST: The expanding world of trinucleotide repeats. Science 271:1374-1375, 1996
- Wiestler OD, Herrlich P: Growth control and neoplastic transformation in the brain. Cancer Cells 3:355-358, 1991
- Wolman SR: Karyotypic progression in human tumors. Cancer Metast Rev 2:257-293, 1983