
Translational Impact of Basic Research Studies of Nicotinic Acetylcholine Receptors
Ronald J. Lukas, PhD
Laboratory of Neurochemistry, Division of Neurobiology, Barrow Neurological Institute, St. Joseph’s Hospital and Medical Center, Phoenix, Arizona
*Courtesy of Ronald J. Lukas, PhD
The Lukas Laboratory
Ronald J. Lukas obtained his PhD in biophysics for work on molecular mechanisms of muscle contraction at the State University of New York Health Sciences Center in Brooklyn after initiating doctoral work in physics at Columbia University in Manhattan. His postdoctoral work was on nicotinic acetylcholine receptors with Edward L. Bennett in the Melvin Calvin Laboratory of Chemical Biodynamics and the Lawrence Berkeley Laboratory at the University of California, Berkeley, and on nerve growth factor with Eric Shooter in the Department of Neurobiology at Stanford University School of Medicine. Yen-Ping Kuo obtained her BS in medical technology at the National Taiwan University, MS in clinical laboratory science at Michigan State University, and PhD in microbiology and molecular biology at Ohio State University for work on archaea. Allan W. Scruggs has a BS in biomedical/biochemical engineering from the University of Southern California and a PhD in biochemistry from Arizona State University where he studied engineered fluorescent proteins. Marsha Segerberg has a BS in biology from the University of Cincinnati and a PhD in biology from the University of Wisconsin for electrophysiological studies. Lori M. Buhlman has a BS in psychology from Michigan State University and is a neuroscience PhD candidate at the University of Arizona. Teresa A. Murray has a BSE in biomedical engineering and is a biomedical engineering PhD candidate at Arizona State University. Linda Lucero obtained her BS in microbiology from the University of Texas at El Paso and an MS in microbiology and immunology from the University of Colorado Health Sciences Center. Kari A. Lindenberger has bachelor degrees in biochemistry and French from the University of Kansas and an MS in biomedical sciences from the Mayo Clinic in Rochester. J. Brek Eaton holds a BS in biology from Arizona State University. Syndia K. Marxer-Miller has a BS in life sciences from Arizona State University West. Jennifer Stevens holds a BS in molecular biology from the University of Arizona. Mercedeh Saba, who has a BS in biochemistry from the State University of New York at Stony Brook, and Kim Walker, who holds a BS in microbiology from Arizona State University, work in the Clinical Assay Development Laboratory overseen by Dr. Lukas and also participate in research studies requiring analysis using immunoassays.
Studies of chemical signaling mediated by the neurotransmitter, acetylcholine, and its receptor targets, including nicotinic acetylcholine receptors, date from the 1850s. The current concepts about chemical transmission and receptive substances came from insights gained early in the 20th century. More recent discoveries reveal a rich diversity in cholinergic receptors and means of cholinergic signaling that present research challenges but therapeutic opportunities. Current nicotinic receptor research is providing tangible insights into the molecular and cellular mechanisms involved in many maladies including nicotine dependence, Alzheimer’s and Parkinson’s diseases, schizophrenia, depression, and attention deficit disorder. This article illustrates how basic nicotinic receptor research informs and is informed by clinical studies and how it serves as a model for contemporary, translational, and biomedical research.
Key Words: acetylcholine, nicotine, receptors, transmitters
Abbreviations used: ACh, acetylcholine; cDNA, copy DNA; DNA, deoxyribonucleic acid; GABA, gamma amino butyric acid; MPTP, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; mRNA, messenger RNA; nAChR, nicotinic ACh receptors; RNA, ribonucleic acid
Much of the current understanding about synaptic neurotransmission is based on studies of chemical signaling through ACh.[15,16,18] ACh is stored in syn- aptic vesicles and released from electrically active nerve terminals. Actions on the postsynaptic target of ACh that are mimicked by the mushroom alkaloid, muscarine (see Figure 1 in Lukas[15]), and those that are particularly sensitive to blockade by atropine are mediated by muscarinic ACh receptors. These receptors are prototypes of the extensive, G-protein-coupled family of metabotropic neurotransmitter receptors.[15]
Muscarinic ACh receptors are composed of monomers or dimers of integral membrane proteins containing seven transmembrane domains. The signals that they mediate affect intracellular levels of second-messengers (e.g., cyclic nucleotides, inositol phosphates, calcium ions, depending on the receptor, and interacting G-proteins). Subsequent effects can include changes in protein kinase activities, cellular metabolism, and gene expression.
Postsynaptic actions of ACh that are mimicked by the tobacco alkaloid, nicotine (see Figure 1 in Lukas[15]), are mediated by nAChR. These receptors are prototypes of the superfamily of neurotransmitter-gated ion channels (ionotropic neurotransmitter receptors).[15,16,18] Depending on the specific neurotransmitter, the binding of chemical messengers “gates” or causes channels to open, increasing permeability to sodium or chloride ions. Electrical activity in the postsynaptic cell is then enhanced or suppressed, respectively.
In the brain, simultaneous inputs through chemical messengers are integrated across neuronal soma and dendritic trees. If the excitatory-to-inhibitory ratio and the absolute level of excitatory signals reach a certain threshold, the neuron becomes electrically active and completes the next segment in the neuronal circuit.
Distribution and Function of Nicotinic AChR
nAChR are broadly distributed across the brain and body. By serving as ligand-gated cation channels,[2,3] they mediate classic excitatory neurotransmission in response to ACh. Subsynaptic to motor neuronal terminals, the activation of nAChR by synaptically released ACh makes movement possible by allowing sodium ions to enter muscle cells. The resulting localized depolarization of the muscle cell membrane potential ultimately triggers the intracellular release of calcium and activation of the contractile apparatus.
Fight-or-flight responses, homeostatic mechanisms, and subconscious control of bodily functions through the autonomic nervous system require the release of ACh from preganglionic nerve terminals and stimulation of postganglionic neuronal electrical activity through nAChR. Evidence continues to emerge that nAChR also play roles in classic excitatory neurotransmission in the brain and spinal cord. These receptors thus contribute to perception, emotion, and cognition, in part as revealed by reports of individuals who use tobacco products and are thus exposed to and experience the effects of nicotine on the brain. Evidence also supports the expression of nAChR in the enteric nervous system, on pulmonary neuroendocrine cells, on tendon fibroblasts, on keratinocytes, in lymphocytes and in thymic stromal elements of the immune system, in the adrenal gland, in the microvascular system, in the prostate gland, in the testes, and in spermatozoa. The presence of nAChR in the cochlea, retina, and olfactory system indicates prominent roles in sensation. Little work has been done on nAChR in these tissues. However, new roles for nAChR, and perhaps explanations for the effects of smoking on them, will likely be revealed by expanded investigations.
Studies in the nervous system as well as in the other tissues mentioned indicate novel roles for nAChR beyond mediation of classic neurotransmission.[16,18] For example, nAChR on or near nerve terminals may modulate the release of other neurotransmitters, including GABA, glutamate, dopamine, serotonin, norepinephrine, and ACh itself. In addition to their function as ACh-gated sodium ion channels, many nAChR have significant calcium ion permeability. The entry of calcium ions into nerve terminals is the signal that stimulates neurotransmitter release. Calcium ion-permeable, presynaptic nAChR thus are positioned and equipped to modulate neurotransmitter release, perhaps even in the absence of an action potential arriving at those nerve terminals.
Cholinergic signaling through nAChR can change synaptic contacts at many stages of life. Depending on factors that remain poorly understood, ACh and nicotine can either attract or deflect neuronal growth cones. This function, however, indicates how dendritic and perhaps axonal architecture can be influenced by nicotinic cholinergic signals. Relevant effects include influences on glutamatergic input to the auditory cortex during a critical phase of development.[23] Thus, throughout the life span, nAChR participate in completing neuronal electrical circuits; influence where, when, and how the substrates for that circuitry are established; and affect the chemical soup in which that circuitry is bathed.
Clearly, nAChR play many critical roles in the nervous system. If something goes wrong with nAChR and nicotinic cholinergic signaling, pathology is a likely consequence. nAChR also are ideally situated to serve as targets for the manipulation of brain and body functions. There are many examples of how the actions at nAChR of toxins or of recreational or medicinal drugs influence organisms negatively or positively.
Diversity of Structure and Function of nAChR
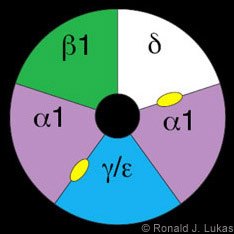
The classic prototype of a ligand-gated ion channel is the muscle-type nAChR (Fig. 1).[16,18] It is a heteropentamer of nAChR building blocks or “subunits,” each of which is encoded by a different gene with a unique chromosomal location. The muscle-type receptor subunits are named alpha 1, beta 1, delta and either gamma (found in the fetal form of mammalian muscle-type receptors) or epsilon (found in the adult form).[19] The subunits of the muscle-type nAChR assemble like staves of a barrel to create a central void, part of which is the ion channel. Ligand binding is the initial step in a process that leads to channel opening.
Each nAChR subunit is a transmembrane protein with the following topology. (1) An extensive, N-terminal, extracellular domain contains sites for ligand recognition and for inter-subunit interactions critical to the formation of the closed assembly. (2) Four transmembrane domains not only anchor the receptor into the membrane but also either line the channel per se (the second transmembrane domain) or form the outer “crust” of the transmembrane region (first, third and fourth transmembrane domains). (3) A short, intracellular segment is between the first and second transmembrane domains. (4) A short, extracellular segment between the second and third transmembrane domains may participate in transduction of ligand binding to channel opening. (5) A short, extracellular, C-terminal ending follows the fourth transmembrane domain. (6) A large, intracellular or cytoplasmic loop is between the third and fourth transmembrane domains (see Model 1 in Figure 5 of Lukas[15]).
The large, N-terminal, extracellular domain, the four transmembrane domains, and the regions of the large cytoplasmic loop nearest or proximal to the third and fourth transmembrane domains (which may form five radiating tunnels that could serve as cytoplasmic extensions of the central transmembrane pore) have amino acid sequences that are highly conserved across all known mammalian nAChR subunits. However, the remaining, “nested” cytoplasmic loop sequences are unique for each nAChR subunit and thus serve as a distinguishing fingerprint.
From a basic and diagnostic laboratory perspective, this fingerprint is important. Antibodies specifically targeting these sequences can be used to identify and discriminate nAChR subunit building blocks. Moreover, oligonucleotide or RNA probes for nAChR subunit mRNA or gene DNA and oligonucleotide primers needed for amplification via the reverse transcription-polymerase chain reaction of subunit message or genes can be designed to be specific for each nAChR subunit.
Functionally, the unique amino acid sequences of cytoplasmic loops provide substrates that permit the following. First, differential trafficking of nAChR subunits to distinct parts of the cell (i.e., dendrites, soma, axons) can occur. Second, differential posttranslational modification of subunits, for example, by phosphorylation, is possible with consequences for receptor assembly, trafficking, and function. Third, differential interaction with cytoskeletal and cytoplasmic elements potentially allows subunit-specificity in trafficking and signal transduction through other intracellular pathways.
Initial protein chemical studies of nAChR focused on receptor-rich electric tissue of electric eels and rays, which have 100 to 1000 times more receptors than innervated mammalian muscle. This work was aided by the ability of toxins from the venoms of poisonous snakes (e.g., cobra, krait, mamba, and sea snake families) to act as probes for nAChR. Like curare, these toxins have the ability to block muscle-type nAChR function by occluding the binding site on receptors for nicotine or ACh.
The toxins are proteins composed of about 72 amino acids. They can be immobilized to allow the capture of receptors. They can be radiolabeled to allow receptors to be counted while they are purified and then studied to determine if other drugs can prevent toxin-receptor interactions. This approach defines the pharmacological profile of drugs acting at nAChR. Finally, the toxins can be tagged with enzymes or fluorophores to permit localization of nAChR at cellular or subcellular levels in different tissues.
Once muscle-type nAChR were isolated, the N-terminal amino acid sequence of each subunit was determined. This work set the stage for the creation of degenerate oligonucleotides that were used as initial probes to identify, clone, and sequence complementary or cDNA corresponding to genes and mRNAs encoding each subunit. For almost a century, nAChR in muscle have been known to have different properties than nAChR elsewhere in the body. However, the bases for those differences had been elusive.
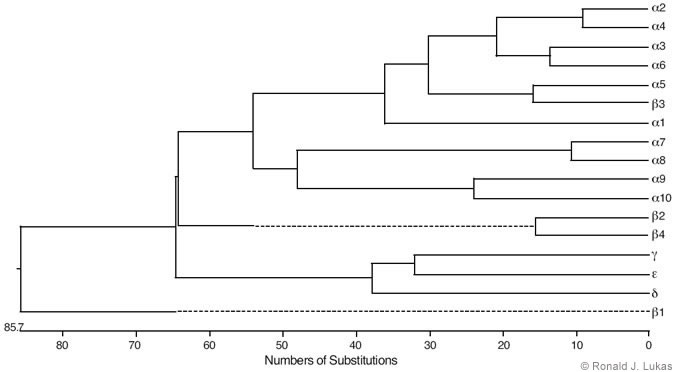
Low-stringency hybridization of muscle-type nAChR subunit cDNA sequences with entities in cDNA libraries from brain and other tissues led to the identification of a family of nAChR subunits from vertebrates that now include alpha 1 through alpha 10, beta 1 through beta 4, gamma, delta, and epsilon subunits.[3,19] Some of these subunits are more alike than others, and the estimated evolutionary distances between them indicate an initial divergence from a primordial gene (Fig. 2). The vertebrate nAChR subunit tree branched with regularity, and the most recently evolved are the alpha 2/alpha 4, alpha 3/alpha 6, alpha 5/beta 3 and beta 2/beta 4 pairs.
{kind=link}
Thus, the long-identified diversity in nAChR subtypes could be explained, at least partially, by the diversity in the genes that encode nAChR building blocks or subunits. By definition, the diverse family of nAChR subtypes bind nicotine and ACh and function as ligand-gated ion channels. However, the distinctive profiles for drug interactions, kinetics for channel opening and closing, and tissue-specific and subcellular locations of each nAChR subtype partially reflect the diversity in the composition of their subunits.
Formally, there is the possibility that there are just under 17[5] possible nAChR subtypes (five positions per pentamer, 17 possible subunits per position, but some with degenerate outcomes). However, some rules seem to dictate the kinds of nAChR subtypes that are assembled (Fig. 3).[3,18,19] Much of this understanding has been established using frog oocyte expression systems. For a given set of subunits, RNA or cDNA is injected into the cytoplasm or nucleus, respectively, of the physically large eggs. The transcription and translation functions of the oocytes can then be hijacked, and success in forming ligand-binding and functional nAChR can be evaluated.
Oocyte expression studies indicate that the muscle-type nAChR subunits are not found in any other assemblies, consistent with tissue studies performed to date. To some degree, in the comparatively liberal oocyte expression system, beta 2 and beta 4 subunits can substitute for beta 1 subunits in assemblies with the other muscle-type nAChR subunits. However, this substitution does not seem to happen naturally. Thus, alpha 2 through alpha 10 and beta 2 through beta b4 subunits (alpha subunits are distinguished from the others because they have relatively rare tandem cysteine residues at about the same location in their large extracellular domains) are the prime candidates for the formation of nAChR found in neurons and perhaps other tissues.
Among the phylogenetically most ancient of these subunits are alpha 7, alpha 8 (found in chickens but not yet in mammals), and alpha 9. In the oocyte expression system, these subunits have the capacity to form homo-oligomers, that is, a receptor where each position in the pentamer is occupied by the same kind of subunit. In the chick, there is evidence for homomeric alpha 7- and homomeric alpha 8-nAChR. There is also evidence for co-assembly of these subunits, although there seems to be no apparent alpha 8 subunit in mammals, precluding the existence of mammalian alpha 7 alpha 8-nAChR.
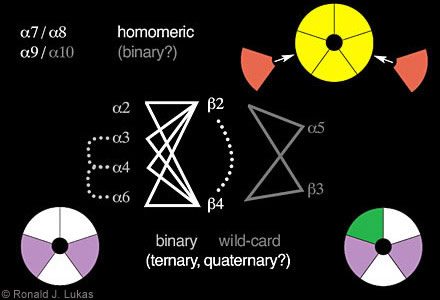
(Figure 3. Schematic diagram indicates some of the rules for nAChR subunit combinations. The subunits alpha 7, alpha 8 and alpha 9 have the capacity to assemble as homomers. In the chick, however, alpha 7 and alpha 8 can exist in the same assembly. Alpha 10 can combine in binary fashion with alpha 9 subunits to form a more highly functional nAChR than created by alpha 9 subunits alone. Other binary complexes can be formed containing alpha 2, alpha 3, alpha 4, or alpha 6 subunits with beta 2 or beta 4 subunits into which alpha 5 and beta 3 subunit “wild-cards” can also integrate to form ternary complexes. Evidence also indicates (dashed lines) that nAChR can contain alpha 3 plus alpha 4, alpha 3 plus alpha 6, or alpha 4 plus alpha 6 subunits or beta 2 plus beta 4 subunits, thus forming quaternary or even a heteropentamer with five different kinds of subunits. The color coding suggests possible positions of subunits in closed assemblies.)
Although alpha 9 subunits can form homomers, levels of functional expression are enhanced dramatically when co-expressed with alpha 10 subunits. This finding suggests binary complex formation in which one or more positions in the pentamer can be occupied by different subunits. Binary complex formation seems to be a rule for the assembly of nAChR containing alpha 2, alpha 3, alpha 4 or alpha 6 subunits in pairwise fashion with either beta 2 or beta 4 subunits. None of these subunits can efficiently form functional nAChR on their own.
nAChR alpha 5 and beta 3 subunits seem to be wild cards. They do not form functional receptors either alone or in pairwise or binary combination with any other subunits. However, alpha 5 and beta 3 subunits can integrate into binary alpha 2/ alpha 3/ alpha 4/ alpha 6- beta 2/ beta 4-nAChR to form ternary complexes with novel functional and ligand-binding properties. Moreover, some functional nAChR can contain both beta 2 and beta 4 subunits, and co-assembly has been observed for alpha 3- alpha 4, alpha 3- alpha 6 and alpha 4- alpha 6 pairs. That is, other ternary and quaternary nAChR can be formed, and perhaps each position in the pentamer could be occupied by a different subunit.
In any event, muscle-type nAChR a1 and non- alpha 1 family genes diverged through a process that culminated in the emergence of delta and then of the gamma/epsilon subunits to allow formation of a quaternary complex. Nonetheless, nonmuscle-type nAChR composed of comparatively newer building blocks but with comparable structural complexity can exist.
The existence of different genes encoding these subunits also allows a high degree of flexibility in controlling the expression of nAChR subunits, which must have an evolutionary advantage. The promoters and enhancers for these genes are unique. Epigenetic influences impinging on cells help dictate which of the cadre of nAChR subunit genes is expressed and when.
This process can have developmentally relevant consequences. For example, muscle-type nAChR containing gamma subunits from low-conductance channels that are open for a long time are ideal for sensing the arrival of developing ACh-releasing motor nerve terminals. In comparison, innervated muscle nAChR containing epsilon subunits form channels that open and close more quickly but that gate larger amounts of current. Such structures are ideally suited to control sequences of muscle contraction in response to rapid pulses of neurotransmitter release.
Thus, nAChR are diverse. Different nAChR subtypes are classified by the kinds of their constituent subunits and the stoichiometries between them. Aside from muscle-type nAChR, a prominent nAChR subtype found in autonomic ganglia contains alpha 3 and beta 4 subunits. nAChR composed of alpha 7 subunits as homomers are prominent in the brain and autonomic neurons. The most abundant brain nAChR with a high affinity for nicotine is composed of alpha 4 and beta 2 subunits. nAChR alpha 3 and alpha 6 subunits are richest in midbrain dopamine neurons.
When at least one constituent subunit in an nAChR subtype is known, but there are or may be others, an “*”is used to designate such a possibility.[19] For example, ganglionic nAChR known to contain alpha 3 or alpha 3 and beta 4 subunits but that also may have other assembly partners are called alpha 3*- or alpha 3 beta 4*-nAChR. Before our understanding of these signaling molecules is complete, much more work is needed to define native nAChR subtypes; their tissue, cellular, and subcellular distributions; the consequences of their activation or blockade; and how these parameters change during development and aging.
The continuing work of Unwin and colleagues has provided tremendous insight into the structure and function of nAChR. They used X-ray scattering from electron microscopic images of tubular arrays of electric tissue nAChR.33 X-ray crystallography has been used to study a snail glial cell ACh-binding protein.[1] The latter is a homologue of a nAChR homopentameric assembly of large, extracellular domains. Its structure reveals important features of nAChR, including their ligand-binding domains, and it is amazingly consistent with findings from site-directed mutagenesis studies of muscle and other nAChR subtypes.8
Dr. Robert D. Scavetta created and I annotated a homology model based on the ACh-binding protein subunit atomic coordinates, which were overlayed with human nAChR a7 subunit N-terminal extracellular domain sequences (Figs. 4 and 5). When viewed from below (i.e., from the plasma membrane, Fig. 4), a clockwise arrangement of features is evident for each subunit: (1) the C-terminal end of the extracellular domain as it transitions to the first cytoplamsic loop, (2) the tip of the so-called “cystine loop,” (3) the disulfide link between cysteine residues that defines the cystine loop, and (4) the tandem cysteine disulfide. The cystine loop is a feature preserved across all members of the four-transmembrane domain superfamily of neurotransmitter-gated ion channels, including nAChR and ionotropic GABAA, glycine, and serotonin-3 receptors. This tip seems to be positioned to exert close communication with the transmembrane segments of the receptor. The tandem cysteine disulfide is known to be in the ligand-binding domain and overlaps spatially with segments of the neighboring subunit at the interface between them.
When viewed from the side (Fig. 5), each subunit resembles an element in an interwoven basket. Anti-parallel beta-pleated sheets represent bundled threads crossing each other to form the interwoven torso of the subunit. At the ends of each assembly are loops of different lengths. The cystine loop ends near the plasma membrane region and just clockwise (viewed from below) from the C-terminus of the same subunit. Another loop ends at the tandem cysteine residues and forms a wingtip overlaying sequences from the neighboring subunit. A third, more extracellularly apposed, extended loop is called the “main immunogenic region”[13] because it is targeted by antibodies. These antibodies recognize complexes of muscle-type nAChRcoupled through the ligand-binding domains with curaremimetic neurotoxins. The main immunogenic region is located in each subunit near the a-helical structure that becomes the N-terminal region of the domain after removal of the signal sequence. During translation on the ribosome, the signal sequence specifies the transit of the protein extracellular domain through to the endoplasmic reticulum lumen for eventual extracellular exposure.
When viewed from below, just counterclockwise from the tandem cysteine loop are two other loops. These loops contain residues on their tips that also influence ligand binding. On the opposing face of the neighboring subunit, residues at the tips of two loops just clockwise from the main immunogenic region also influence ligand binding. Thus, interfaces between subunits provide points of contact and recognition that are essential to the formation of the closed, pentameric assembly. They are also points for ligand recognition.
ACh and nicotine are thought to bind at subunit interfaces (Fig. 1, yellow ovals). It is easy to imagine how those ligands can gain access to the binding pocket between subunits and behind the tandem cysteine loop. The wingtip would then be pushed away from the axial center of the structure. This push would cause the interwoven torso, including the cysteine loop, and the attached (but not illustrated in these images) second transmembrane domain to rotate, leading to channel opening.
Relevance to Health and Disease
Our work has focused on the use of cell lines as models for studies of human nAChR.[17,20] These cell lines are from human tumors and hence are immortalized. Thus, they can be propagated to achieve almost any experimental end. They can be used for pharmacological studies to define nAChR subtypes based on their drug-binding properties gleaned from radioligand binding or functional studies. Some of these cell lines, such as rhabdomyosarcomas and peripheral neuroblastomas, naturally express the same nAChR subtypes found in analogous, non-neoplastic tissues, namely, muscle and autonomic neurons, respectively.
However, many important nAChR subtypes, especially those expressed in central neurons, are not naturally expressed by a known cell line. Consequently, we used genetic-engineering techniques to create new cell lines that heterologously express nAChR composed as combinations of specific subunits. We have been most successful with the SH-EP1 human epithelial cell line as a host for stable transfection with cDNAs encoding select combinations of nAChR subunits. This line does not naturally express any nAChR subunits or subtypes. Stably transfected SH-EP1 cells thus represent a complement to the frog oocyte system for heterologous expression of nAChR subtypes of interest.
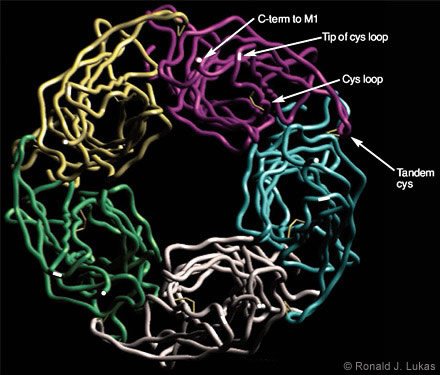
and (4) the tip of the loop containing the tandem cysteine residues, which form another di- sulfide link at the ligand-binding site.)
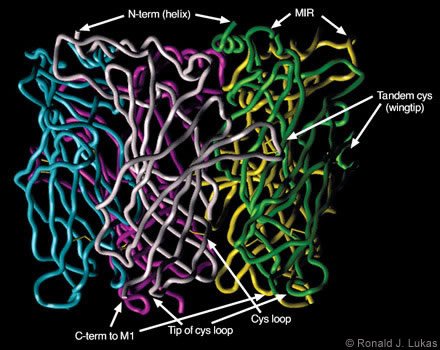
function of nAChR, the design of drugs that would interact with them, and antigen selection for the generation of useful antibodies.)
Using these cells, we can help define the realm of possibilities for nAChR subunit composition. Information gleaned from this work expedites determination of the makeup of naturally expressed nAChR subtypes. We also can define the functional and ligand-binding pharmacological profiles of specific nAChR subtypes. The profiles provide a means of discriminating between nAChR subtypes when they are naturally expressed. They are also a resource for identifying and refining nAChR subtype-specific (in absolute terms) or subtype-selective (compared to interactions with other nAChR subtypes) drugs that can be used for research or as prototypes for clinical and medicinal use. For example, we have helped to define the selectivity of dihydro-beta-erythroidine for human alpha 4 beta 2- over alpha 3*-nAChR and of methyllycaconitine for alpha 7-nAChR.[2,21,35]
In site-directed mutagenesis studies, cDNA mutations are created to produce nAChR subunit proteins with altered amino acids or interest. In chimeric receptor studies, cDNAs are engineered to allow strings of amino acids to be altered. Such studies have been used to define structure-function relationships and as models for disease. For example, chimeric studies have revealed unexpected roles of nested cytoplasmic loop sequences in alpha 4 beta 2-nAChR function and essential roles for proximal cytoplasmic loop sequences for expression of alpha 4beta 2-nAChR on the cell surface.[9]
The numbers of nAChR in the brains of patients with Parkinson’s or Alzheimer’s disease are lower than in the brains of age-matched controls. In Alzheimer’s disease those losses precede all other hallmarks of the disease.[27] The incidence of Parkinson’s disease in smokers is 50% less than it is in nonsmokers. In animal, tissue slice, or cell-culture models, nicotine exposure can protect against the neurotoxic effects of beta-amyloid, a suspected etiopathogenic agent in Alzheimer’s disease. It also protects against the effects of MPTP, a recreational drug that causes Parkinson’s-like symptoms in users. Moreover, nicotine is useful as a procognitive drug in patients with Alzheimer’s or dementia related to Parkinson’s disease.
Neurons that stain positively for immunoreactive nAChR subunits in control postmortem human brains are lost in Alzheimer’s disease. Instead, the senile plaques and neurofibrillary tangles associated with later-stage Alzheimer’s disease stain for nAChR subunits. These findings may imply defects in the trafficking of nAChR at some stage of the disease.[31] Serendipitously, we also noted nAChR subunit-like immunoreactivity in the brain microvasculature, which has stimulated work along those lines. Furthermore, at the concentrations found in diseased brains, beta-amyloid can act acutely to inhibit the functional responses of nAChR to nicotine or ACh. This finding suggests a possible etiopathogenic role for receptor block in disease onset, progression, or both.[34]
An improved understanding of the effects of chronic nicotine exposure on brain and body function is relevant to smoking behavior and public policy to help control or end the behavior. However, it is also relevant to the prospective medicinal use of nicotinic drugs. A subset of the American adult population is chemically dependent on nicotine. A much larger proportion of the adult populations in the Pacific rim and eastern Europe in particular are likewise dependent. This dependence drives smoking and other tobacco-use behaviors. It is likely that interactions at nAChR contribute prominently to nicotine dependence.
Our work using cell line models indicates that every nAChR subtype tested to date becomes initially activated when transiently exposed to nicotine.[6] However, sustained exposure leads to inactivation of function through an apparent series of processes called “desensitization” and “persistent inactivation.” These effects occur with nicotine concentration and time-of-exposure dependencies that differ for each nAChR subtype. However, upon persistent exposure to nicotine at concentrations like those found in the plasma of smokers, numerically abundant alpha 4 beta 2-nAChR function to only 50% of their capacity.[7]
Thus, we hypothesize that smokers learn to self-administer nicotine to levels that produce a certain level of nAChR functional blockade. In this regard, nicotine is far different from drugs of abuse such as cocaine or heroin or amphetamine, which are delivered in a way to stimulate their targets. Our findings also suggest that drugs that partially block or only partially stimulate select nAChR subtypes would have physiological consequences similar to those associated with the use of tobacco products and recreational nicotine. Such drugs might serve as effective substitutes for smoking.
That idea is supported by the success of smoking cessation clinics based on the use of bupropion (Wellbutrin, GlaxoSmithKline, United Kingdom; Zyban, GlaxoSmithKline, United Kingdom). When present at clinically effective doses, this compound is an nAChR blocker.[5] Nicotine-mecamylamine treatment[26] also would produce an nAChR-selective block. The recent announcement of the development of varenicline as a new aid to smoking cessation underscores this idea. This drug is a “partial agonist” (an agent that partially activates) selective for human a4b2-nAChR.[28]
As many as 90% of schizophrenics are smokers.[11,12] These individuals tend to report that their moments of tranquility occur during smoking. We have conducted studies indicating that a number of clinically useful antipsychotic drugs inhibit the function of nAChR at medically achieved concentrations. These findings again support the idea that inhibition of nAChR function by nicotine exposure is central to the self-medicating effects of tobacco products.[22] Roles for nAChR in normal functions affected in psychosis also are suggested.
About 90% of alcoholics are smokers. There are suggestions that hangovers and loss of cognitive function are moderated by such nicotine exposure, but the relationships between the effects of nicotine and ethanol remain elusive.
Recent epidemiological studies suggest that the risk for smoking behavior in teens is modest for offspring of smoking parents, slightly higher for siblings of smokers, but higher yet for individuals whose peers smoke.[32] Individuals with a history of depression or anxiety have an even higher risk for smoking.[32] A history of attention deficit problems is associated with the highest risk of smoking.[32] Some users report that nicotine stimulates them, whereas others report that nicotine calms them. These reports are not contradictory when it is realized that nicotine is a state-dependent mood stabilizer that calms those who are anxious and elevates moods in those who are depressed.[29]
We postulate that individuals who are susceptible to nicotine dependence have heightened nicotinic cholinergic signaling, which is perhaps associated with mood instability and attentional difficulties.[6] As nicotine exposure begins to lower nAChR activity and their hyperactive nicotinic cholinergic signaling is dampened, such individuals may begin to sense (probably subconsciously) their more balanced mood and their enhanced attentional and consequent cognitive performance. Our studies suggesting that tobacco use represents self-medication to treat underlying emotional and cognitive problems are supported not only by the risk studies mentioned above, but also by data indicating that about 40% of adult smokers in the United States are currently being or recently have been treated for depression, anxiety, or attentional problems.[11] What if the rest of the smokers also had the same issues, but subclinically? Or what if they have assumed life styles that keep them out of clinics? Our work showing that various antidepressants block the function of nAChR subtypes supports the “nicotine self-medication to produce nAChR inactivation” hypothesis.[4]
Collectively, our studies lead us to suggest that smoking cessation will be successful when nicotine dependence is treated successfully (e.g., by providing enough nicotine through patches or other devices to yield blood and brain levels equal to those achieved by the individual while smoking). However, success in smoking cessation also requires treatment of underlying psychiatric issues.
Myasthenia gravis is a neuromuscular disease caused by an autoimmune response against muscle-type nAChR. The mechanism was discovered serendipitously when scientists sought to generate antibodies against electric fish nAChR. They found that the inoculated rabbits developed a characteristic, flaccid paralysis that was reversible with conventional anticholinesterase therapy.[25] Other myasthenic syndromes are caused by specific mutations in muscle-type nAChR subunits.[14] Similarly, some forms of idiopathic epilepsy are caused, not by defects in inhibitory chemical neurotransmission, but by mutations in brain nAChR subunits. This is another arena where heterologous expression studies and site-directed mutagenesis work are providing insight into disease states. Serendipity also intervened in observations that nicotine exposure lowered the frequency of tics in children who were depressed and suffering from Tourette’s syndrome, thus providing insights into roles of nAChR in that behavioral disorder.[30] Autoimmune reactivity and mutations in nAChR subunits found in keratinocytes and perhaps other skin cell types also have been implicated in the skin disorder, acantholysis.[14]
We have extended our studies to other tissues and organ systems, mostly through collaborations. We have used our cell lines as models for the development and refinement of techniques needed for histological studies; for the detection of nAChR subunit messages, proteins, and binding sites at low levels; and for localization of those entities with cellular or subcellular resolution. These studies have concerned native nAChR in the brain’s pleasure-reward centers relevant to nicotine dependence and its effects on mood and emotion. Studies in the spinal cord have focused on acute and chronic pain (one compound from the skin of a type of South American frog has 100 to 1000 times more analgesic potency than morphine; rather than being targeted at opioid receptors, it is a powerful activator of nAChR function).
These studies have also involved nAChR and their influences in the immune system. Organ cultures using thymus from nonobese diabetic, severely combined immunodeficient mice that lack hemopoietic components of the immune system recombined with sources rich in those cells (e.g., mouse spleen or liver or human umbilical cord blood cells) have been used to mimic events in the early development of the immune system. We have found widespread expression of nAChR in both stromal and hemopoietic components of the cultures, that nicotine exposure perturbs thymic signaling, and effects of prenatal nicotine exposure on the fate of cord blood cells from smoking mother-fetus pairs. These findings have implications for the effects of nicotine exposure on the early development of the human immune system.[10,24]
Conclusions
Collectively, this is a tale of discovery and insight concerning nAChR, spun from studies of electric fish, a drug from the tobacco plant, toxins from frogs and snakes, nAChR homologues from snails, and the ingenuity of investigators riding the crest of waves of technical development ranging from drug studies to recombinant DNA and genetic engineering. It beautifully illustrates how discoveries from diverse lines of investigation couple with serendipity and interpretive imagination to reveal secrets of one of nature’s most intriguing and important class of molecules. Now, gene knock-in and knock-outs, application of techniques such as single ion-channel studies and creation of monoclonal antibodies, and powers of combinatorial chemistry and intelligent drug design are converging to provide tools and approaches that are allowing nAChR to be studied, even when present at low levels and in complex mixtures. This work is moving forward using a variety of animal models as well as with regenerable and postmortem human tissues. The findings are providing greater insights into nAChR and their normal and pathophysiological roles.
Acknowledgments
Thanks are due to Dr. Robert D. Scavetta of the Mayo Clinic, Scottsdale, for his timely work to generate the nAChR alpha 7 extracellular domain homology model so useful in visualizing features of the entire class of molecules. The author also is indebted to his colleagues in the Laboratory of Neurochemistry and to many valued collaborators who have participated in the work mentioned.
References
- Brejc K, van Dijk WJ, Klaassen RV, et al: Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 411:269-276, 2001
- Eaton JB, Peng JH, Schroeder KM, et al: Characterization of human alpha 4 beta 2-nicotinic acetylcholine receptors stably and heterologously expressed in native nicotinic receptor-null SH-EP1 human epithelial cells. Mol Pharmacol 64:1283-1294, 2003
- Elgoyhen AB, Vetter DE, Katz E, et al: Alpha10: A determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc Natl Acad Sci USA 98:3501-3506, 2001
- Fryer JD, Lukas RJ: Antidepressants noncompetitively inhibit nicotinic acetylcholine receptor function. J Neurochem 72:1117-1124, 1999
- Fryer JD, Lukas RJ: Noncompetitive functional inhibition at diverse, human nicotinic acetylcholine receptor subtypes by bupropion, phencyclidine, and ibogaine. J Pharmacol Exp Ther 288:88-92, 1999
- Gentry CL, Lukas RJ: Regulation of nicotinic acetylcholine receptor numbers and function by chronic nicotine exposure. Curr Drug Targets CNS Neurol Disord 1:359-385, 2002
- Gentry CL, Wilkins LH, Jr., Lukas RJ: Effects of prolonged nicotinic ligand exposure on function of heterologously expressed, human alpha4beta2- and alpha4beta4-nicotinic acetylcholine receptors. J Pharmacol Exp Ther 304:206-216, 2003
- Grutter T, Changeux JP: Nicotinic receptors in wonderland. Trends Biochem Sci 26:459-463, 2001
- Kuo Y-P, Xu L, Eaton JB, et al: Roles for nicotinic acetylcholine receptor subunit large cytoplasmic loop sequences in receptor expression and function. J Pharmacol Exp Ther, in press
- Kuo Y, Lucero L, Michaels J, et al: Differential expression of nicotinic acetylcholine receptor subunits in fetal and neonatal mouse thymus. J Neuroimmunol 130:140-154, 2002
- Leonard S, Adler LE, Benhammou K, et al: Smoking and mental illness. Pharmacol Biochem Behav 70:561-570, 2001
- Leonard S, Bertrand D: Neuronal nicotinic receptors: From structure to function. Nicotine Tob Res 3:203-223, 2001
- Lindstrom J: Neuronal Nicotinic Acetylcholine Receptors in Ion Channels. New York: Plenum Press, 1996
- Lindstrom JM: Nicotinic acetylcholine receptors of muscles and nerves: Comparison of their structures, functional roles, and vulnerability to pathology. Ann N Y Acad Sci 998:41-52, 2003
- Lukas RJ: Neurotransmitter receptor diversity: The nicotinic acetylcholine receptor family. BNI Quarterly 6(2):15-23, 1990
- Lukas RJ: Neuronal nicotinic acetylcholine receptors, in Barrantes FJ (ed): The Nicotinic Acetylcholine Receptor: Current Views and Future Trends. Berlin/Heidelberg: Springer-Vertag and Gerogetown, Texas: Landes Publishing Co, pp 143-156
- Lukas RJ: Cell lines as models for studies of nicotinic acetylcholine receptors, in Arneric SP, Brioni JD (eds): Neuronal Nicotinic Receptors: Pharmacology and Therapeutic Opportunities. New York: Wiley-Liss Inc., 1999, pp 81-97
- Lukas RJ, Bencherif M: Heterogeneity and regulation of nicotinic acetylcholine receptors. Int Rev Neurobiol 34:25-131, 1992
- Lukas RJ, Changeux JP, Le Novere N, et al: International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev 51:397-401, 1999
- Lukas RJ, Fryer JD, Eaton JB, et al: Some methods for studies of nicotinic acetylcholine receptor pharmacology, in Levin ED (ed): Nicotinic Receptors and the Nervous System. Boca Raton: CRC Press, 2002, pp 3-27
- Lukas RJ, Norman SA, Lucero L: Characterization of nicotinic acetylcholine receptors expressed by cells of the SH-SY5Y human neuroblastoma clonal line. Molec Cellular Neurosci 4:1-12, 1993
- Lukas RJ, Wilkins LH: Antipsychotics noncompetitively inhibit function of diverse nicotinic acetylcholine receptor subtypes. J Neurochem 90:37, 2004
- Metherate R, Hsieh CY: Synaptic mechanisms and cholinergic regulation in auditory cortex. Prog Brain Res 145:143-156, 2004
- Middlebrook AJ, Martina C, Chang Y, et al: Effects of nicotine exposure on T cell development in fetal thymus organ culture: arrest of T cell maturation. J Immunol 169:2915-2924, 2002
- Patrick J, Lindstrom J: Autoimmune response to acetylcholine receptor. Science 180:871-872, 1973
- Rose JE, Behm FM, Westman EC: Nicotine-mecamylamine treatment for smoking cessation: The role of pre-cessation therapy. Exp Clin Psychopharmacol 6:331-343, 1998
- Sabbagh MN, Lukas RJ, Sparks DL, et al: The nicotinic acetylcholine receptor, smoking, and Alzheimer’s disease. J Alzheimers Dis 4:317-325, 2002
- Sands SB, Brooks PR, Chambers LK, et al: A new therapy for smoking cessation: Varenicline, a selective nicotinic receptor partial agonist. Proc Soc Res Nicotine Tobacco 11: 14, 2005
- Shytle RD, Silver AA, Lukas RJ, et al: Nicotinic acetylcholine receptors as targets for antidepressants. Mol Psychiatry 7:525-535, 2002
- Silver AA, Shytle RD, Philipp MK, et al: Transdermal nicotine and haloperidol in Tourette’s disorder: A double-blind placebo-controlled study. J Clin Psychiatry 62:707-714, 2001
- Sparks DL, Beach TG, Lukas RJ: Immunohistochemical localization of nicotinic beta2 and alpha4 receptor subunits in normal human brain and individuals with Lewy body and Alzheimer’s disease: Preliminary observations. Neurosci Lett 256:151-154, 1998
- Tercyak KP, Lerman C, Audrain J: Association of attention-deficit/hyperactivity disorder symptoms with levels of cigarette smoking in a community sample of adolescents. J Am Acad Child Adolesc Psychiatry 41:799-805, 2002
- Unwin N: Structure and action of the nicotinic acetylcholine receptor explored by electron microscopy. FEBS Lett 555:91-95, 2003
- Wu J, Kuo YP, George AA, et al: Beta-amyloid directly inhibits human alpha4beta2-nicotinic acetylcholine receptors heterologously expressed in human SH-EP1 cells. J Biol Chem 279:37842-37851, 2004
- Zhao L, Kuo YP, George AA, et al: Functional properties of homomeric, human alpha 7-nicotinic acetylcholine receptors heterologously expressed in the SH-EP1 human epithelial cell line. J Pharmacol Exp Ther 305:1132-1141, 2003