
Genetics of Adult Malignant Gliomas
Joan Rankin Shapiro, PhD
Stephen W. Coons, MD*
Divisions of Neurology and *Neuropathology, Barrow Neurological Institute, St. Joseph’s Hospital and Medical Center, Phoenix, Arizona
Abstract
The genetic aberrations responsible for the formation and progression of malignant gliomas are being identified. It is now possible to correlate some of these aberrations with the specific histopathological and biologic characteristics of astrocytic tumors, oligodendrogliomas, and ependymomas. In defining these progressive alterations, distinctions that are unique to different types of gliomas are being identified as well as potential subsets within specific types of gliomas. This article reviews the genetic changes associated with malignant gliomas. Many of these alterations are being investigated for their potential as diagnostic and prognostic indicators and as new therapeutic targets.
Key Words : astrocytomas, ependymomas, oligodendrogliomas, oncogenes, tumor suppressor genes
Technical advances and hereditary cancers have increased our understanding of the genetic aberrations associated with brain tumors. Genetic cancers have provided insight into the multiple steps required to immortalize a cell, to allow its uncontrolled proliferation, and to invade and modulate the local cellular environment.14,16,32 Brain tumors likewise develop in a multiple step process. From these new findings, models of tumor progression are being developed in which genetic lesions are being associated with specific tumor types and pathological grades.41,84,179
Diffuse Astrocytomas
Diffuse astrocytomas are the most common type of glioma in both adults and children. Adult and pediatric tumors, however, exhibit several notable differences besides histology. The first is a predilection for different locations in the brain. Adult brain tumors tend to develop supratentorially within the cortical, subcortical, and basal ganglia regions. In contrast, about 60% of pediatric tumors develop infratentorially within the cerebellum, pons, and brain stem.147 The incidence of particular tumor types also differs. Astrocytic tumors, oligodendrogliomas, and ependymomas are most common in adults while pilocytic astrocytomas and primitive neuroectodermal tumors (PNET) are more common in children.
The response of adult and pediatric malignancies to therapy also differs.155 Extensive resection, irradiation, and chemotherapy improve survival only modestly in adults with malignant central nervous system (CNS) tumors.154 Pediatric tumors tend to be more responsive to treatment, especially to chemotherapy.56 This differential responsiveness may reflect biological differences in adult and pediatric tumors, even when they appear to be the same histological type and grade. Thus, there is a need to determine if alternative pathways of evolution exist within tumor types and between adults and pediatric brain tumors.
Astrocytomas
Typically, astrocytomas (grade II tumors) occur in individuals between 20 and 40 years old. They tend to grow slowly but are not benign because of their invasive quality and location. Fibrillary astrocytomas can occur anywhere in the brain but are most prevalent in the cerebral hemispheres. Their histology lacks nuclear atypia and mitotic activity. Low-grade tumors are difficult to study because of tissue availability. Needle biopsies are often used to make a diagnosis. Upon resection, these tumors are frequently admixtures of normal reactive cells and tumor cells. The proportion of normal cells to tumor cells can greatly influence experimental results.19,22 The tumors can also appear to be mixtures of different histological grades of tumor. These complexities raise questions about whether low-grade gliomas are cytogenetically normal with molecular genetic lesions, whether specific numerical abnormalities reflect a specific pathway of tumor evolution, or both.84
The cytogenetics of astrocytic tumors is reviewed extensively elsewhere.153 The most common numerical aberration involves the gain of chromosome 7 along with the loss of a single sex chromosome. Structural abnormalities are rare but involve chromosomes 1p and 9p153 when observed.
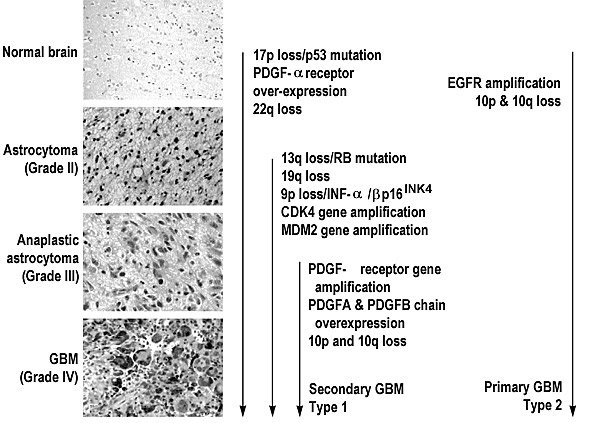
Mutations and allelic loss (loss of one or both genes at a specific locus) have been the primary genetic lesions detected by molecular analyses. One of the early events in the formation of a malignant glioma involves a mutation or allelic loss of chromosome 17p (Fig. 1).35,36,40,59,60,72,86,100,143,156,175,177 The target gene in 17p is the TP53 gene in which more than 200 mutations have been described in human tumors.55 This gene may be important in both initiation and progression.71 Sequence data indicate that a highly conserved region involving exons 5, 7, and 8 bears the majority of missense mutations that inactivate the TP53 gene.85 A brain-specific mutation does not appear to exist. Most DNA lesions in the TP53 gene occur in the so-called “hot spots” involving codons 175, 248, and 273.23 These same abnormalities are associated with most other types of cancer.
The TP53 gene binds to DNA to control the transcription of other genes. Many of its identified mutations cause this function to be lost. The amplification of several oncogenes [e.g., murine double minute 2 (MDM2) or c-myc] also abrogates the function of p53.125,126MDM2 binds to p53 and inactivates it while c-myc enhances its expression.130 p53 protein also affects the expression of growth factors. For example, wild-type TP53 represses the basic fibroblast growth factor (bFGF) transcriptional promoter. This finding is supported by cells with mutated TP53, which exhibit an upregulation of bFGF.169 A similar scenario may exist with vascular endothelial growth factor (VEGF), a mediator of angiogenesis.68 With VEGF these events may be more critical in higher grade tumors because the histopathology of astrocytomas does not support the complete loss of cell cycle control or angiogenesis in lower grade neoplasms.84,190
Mutant TP53 is identified in more than a third of the astrocytomas. It may be important in the genesis of low-grade astrocytomas by abrogating apoptosis or by increasing genomic instability. Of interest is the frequency of TP53 abnormalities in different age groups. TP53 mutations appear to be more common in patients between 18 years and the mid 40s (44%) than in older patients (9%).119,181Consequently, TP53 mutations may follow a different evolutionary pathway in younger patients than in older patients. This phenomenon is discussed in greater detail in the section on glioblastomas multiforme (GBMs).
A second family of genes important in astrocytomas includes the platelet-derived growth factors A and B (PDGFA and PDGFB) and PDGF receptors a and b (PDGFR-a and PDGFR-b) (Fig. 1).191 The PDGFA and B chains dimerize to form AA, BB, or AB homo- or heterodimers. The receptor, PDGFR-a, binds with AA and AB while PDGFR-b binds with BB. In astrocytomas, the A chain and a-receptor are predominantly overexpressed.34,51,52,102 This observation is interesting because the most common chromosomal abnormality identified in astrocytomas involves aneuploidy of chromosome 7,10 the chromosomal location of the PDGF-A chain. In vitro and in vivo, treatment with 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) selects for a minor subpopulation of cells containing amplified PDGF-A and -B chains.145 Changes in the expression of growth factors and their receptors may also initiate local environmental changes that begin to stimulate angiogenesis. Overexpression of PDGFR also correlates with the loss of heterozygosity for 17p.52 Although the exact nature of these events is unknown, PDGF and its receptors clearly play a role in tumor evolution.
In about 30% of the astrocytomas, allelic loss has been identified on chromosome 22q.38,59,99 A likely candidate for this genetic loss was the NF-2 gene, but an extensive analysis of this gene in all grades of gliomas failed to detect a consistent abnormality.54,138Allelic loss on chromosome 22q occurs more telomeric to the NF-2 gene,120,134 but the gene or genes involved have not been identified. Other chromosomes exhibiting allelic loss in astrocytomas include chromosome 1,9,49,174 chromosome 3,61 and chromosome 13.121 Each allelic loss identified in these studies represents a probable site for tumor suppressor genes but awaits confirmation.
Anaplastic Astrocytomas
Anaplastic astrocytomas are thought to develop from low-grade astrocytomas (Fig. 1).15,140 Although no sharply defined histological criteria separate astrocytomas from anaplastic astrocytomas, the latter are distinguished by a high cell density and level of mitotic activity, early or focal contrast enhancement, and a high degree of nuclear polymorphism including multinucleation, lobulation, and angulation.140 The nuclear-cytoplasmic ratio is also increased, but the astrocytic character is still evidenced by fibrillar eosinophilic cytoplasm and the presence of processes.
Anaplastic astrocytomas occur in both young and old patients, but their incidence peaks in the mid50s. Like astrocytomas the gain of chromosome 7 is the most frequent numerical aberration. Chromosomes 19 and 20 also tend to be overrepresented while the chromosomes most frequently lost are chromosomes 10, 22, and a single sex chromosome. The patterns of gain and loss are more prominent in anaplastic astrocytomas as are the numbers of structural abnormalities. Most breakpoints occur on the p arms of chromosomes 1, 3, and 9 (1p32, 1p36, 3p21, 9p21 and 9p22) with similar clusters in the q arms of chromosomes 6 and 7 (6q21 and 7q22) and occasionally in chromosomes 5p13, 15q11, 17p11, and 19q12.1.153
Thirty to forty percent of the anaplastic astrocytomas have a mutation of the TP53 gene (17p13.1) in addition to allelic loss, overexpression of PDGF and PDGF receptors, and allelic loss on 22q.29,35,36,38,59,72,74,86,124,143,156,173,175 Changes that mark the transition from astrocytoma to anaplastic astrocytoma involve allelic loss on 9p, 11p, 13q, and 19q. The changes associated with chromosomes 9 and 13 are important steps in the evolution of this tumor because the target genes are proteins that govern critical steps in the cell cycle.
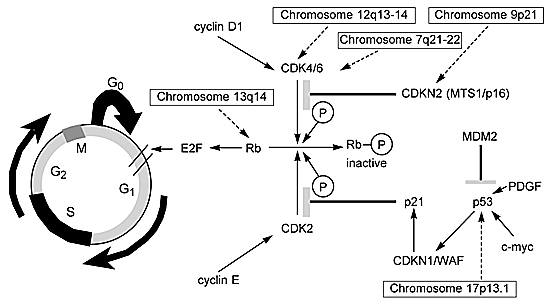
A key gene in this scenario is the retinoblastoma gene (RB) located on chromosome 13q14. The protein encoded by this gene is a checkpoint in the cell cycle that inhibits the transition of the G0/G1®S phase (Fig. 2). When the Rb1 protein is phosphorylated by either CDK4/CDK6 or CDK2, it becomes inactivated and no longer inhibits G1®S. The CDK4/CDK6 and CDK2 genes are controlled by positive (cyclins D1 and E) and negative (CDKN2/p16 or p21 proteins) regulators. The deregulation of either of these genes or genes upstream in the pathway can produce a similar loss of control over the cell cycle.
The function of upstream genes exerting control over CDKN2/p16 and p21 can also be abnormal (Fig. 2). For example, activation of the CDKN1/WAF1 gene, which is under the control of the p53 protein, results in the production of the p21 protein. Wild type p53 protein can be suppressed by the amplification of the oncogene MDM2 or upregulated by c-myc (Fig. 2). Thus, the cascade of events leading to the inhibition of the Rb1 protein is controlled by multiple genes. If any one gene fails to function normally, the cell cycle continues unchecked. In fact, more than one DNA lesion in the critical steps of this cell cycle pathway may be rare.50,170 However, it is possible to use two different markers to determine aberrancy within the cell cycle. For example, comparisons of allelic loss in an inhibitor such as CDKN2/p16 and the proliferative marker Ki-67 demonstrate a distinct relationship. When there is a homozygous deletion of the inhibitor CDKN2/p16, proliferation correspondingly increases.109
Not all deregulation is related to the allelic loss of genetic material. Several investigations have identified a region on chromosome 12 (12q13-14) that is amplified in 15% of World Health Organization grade III (anaplastic astrocytomas) and grade IV (GBMs) tumors.119,126 A number of genes map to this region. Two (i.e., MDM2 and CDK4) are frequently amplified and can deregulate the cell cycle (Fig. 2).25 MDM2 codes for a cellular protein that complexes with the p53 tumor suppressor gene product and inhibits its function. Gliomas, in which TP53 is seldom lost or mutated, can thereby escape TP53-mediated growth regulation.119,126Chromosome 9q21, the map location for CDKN2/p16,63,108 is a region of frequent allelic loss. The loss of this inhibitor104,172 provides an alternative mechanism leading to the same biological endpoint—an increase in CDK4.
Allelic loss on chromosome 19q13.2-13.3 has been observed, although the putative tumor suppressor gene has not yet been identified.135,139,178,180 This gene, which appears to be unique to glial tumors, is the only allelic loss shared by astrocytic tumors, oligodendrogliomas, and oligoastrocytomas (discussed below). Several candidate genes cloned from this chromosome region are under active investigation.171,198
Multiple deletions and rearrangements have also been noted on chromosome 1.153 Analysis of this chromosome has recently discovered a family member of the TP53 gene. Regions spanning 1p36-p32, a site involved in numerous deletions and translocations, demonstrate frequent allelic loss.7,9 Although small, the sampling of anaplastic astrocytomas has detected allelic loss in this region. This finding suggests that the new p73 gene, a gene that is 63% homologous to TP53 in the DNA-binding regions, will function much like TP53.65,66
Other chromosomes identified as having allelic loss include chromosome 11p15®pter39,159 and chromosome 3p21.73 More anaplastic astrocytomas must be analyzed to determine the importance of these findings.
Glioblastomas Multiforme
GBMs are the most malignant of the astrocytic tumors. The mean survival time of patients with this diagnosis is about 1 year (Fig. 1).155 GBMs are highly infiltrative, producing undifferentiated elements as a dominant feature in addition to mitotic activity and necrosis. Vascular proliferation may also be evident, along with a high bromodeoxyuridine/Ki-67 labeling index. Although the genetic instability of this tumor creates numerous and varied genetic changes, malignant astrocytic gliomas develop several nonrandom chromosome changes that are associated with progression.153 The most frequent numerical chromosome changes involve the gain of chromosomes 7 and 20 and the loss of chromosomes 10, 22, and a single sex chromosome as reviewed elsewhere.153 The gain of chromosome 20 is more numerous in GBMs than in anaplastic astrocytomas. In general, numerical changes appear to reflect loss more than gain of chromosomes (e.g., the loss of chromosomes 9, 13 and 14).
GBMs represent about 50% of all intracranial neoplasms. Tissue availability is less of a factor than it is with astrocytomas, and a large amount of molecular data has been generated on this tumor. The frequency of TP53 mutations and/or allelic loss is about the same in GBMs as in anaplastic astrocytomas and astrocytomas. This finding supports the hypothesis that this DNA lesion is an early change in the evolution of gliomas.59 The clonal expansion of a TP53 mutation as the dominant cellular population in a recurrent tumor further supports this hypothesis.156 Another gene independent of TP53, however, may reside at 17p13.3.17
Other significant DNA lesions are associated with the gain of chromosome 7. The genes encoding the epidermal growth factor receptor (EGFR) and the A chain of PDGFA are mapped to chromosome 7 at 7p13-p11 and 7q11-q13, respectively. Both of these genes are amplified or overexpressed in GBMs.10,51 More than half of the GBMs with an amplification of EGFR also have a rearrangement of the gene.27 This mutated form of the EGFR has a high level of tyrosine kinase activity in the absence of the EGF ligand, essentially keeping this receptor in a “turned on” autocrine mode.24 PDGF may be overexpressed less often in GBMs, but its autocrine regulation suggests that like EGFR it could provide a selective growth advantage to tumor cells.
Figure 3 is a hypothetical version of how growth factors and growth factor receptor signaling affect cellular proliferation. Almost every growth factor known to stimulate cell division has been identified as aberrantly expressed in GBM cell lines and fresh tissue.12 Growth factors, such as PDGFA, PDGFB, EGF, transforming growth factor-a and -b (TGF-a and TGF-b), VEGF, insulin-like growth factor (IGF-I and IGF-II), bFGF, and their receptors, are necessary components of an organism during normal development and body function188and in pathology.12 Most of these growth factors have receptor tyrosine kinase activity. The kinase adds phosphate groups to the intracellular domain of the receptor and activates it to initiate the cascade of signals that makes cells divide. The aberrant function of one or more of these factors in gliomas is thought to be one of the major reasons that control of the growth stimulatory pathways is lost. The identification of the tumor suppressor gene (PTEN/MMAC-1, phosphate-tensin/mutated in multiple advanced cancers-1) on chromosome 10q23.3 further strengthens this hypothesis.80,162
Significant loss of the whole chromosome 10 has been found in about 50% of the GBMs analyzed cytogenetically. Allelic loss on chromosome 10p and 10q occurs in as many as 60 to 90% of the gliomas.18,37,67,123 The PTEN/MMAC-1 gene encodes a tyrosine phosphatase, a molecule whose opposing enzymatic activity removes phosphate groups. Among the genes encoding receptor tyrosine kinase activity, two (EGFR and PDGF) are often aberrant in gliomas. EGFR can bind both its ligand EGF and TGF-a (Fig. 3). Because this receptor is the most frequently amplified gene in GBMs,81,195 the amplification-associated overexpression of EGFR could potentially override the normal negative regulation of the PTEN/MMAC-1 gene product.163
In contrast, GBM cells with homozygous deletion, mutation, or both will have an inactivated PTEN/MMAC-1 gene that will completely eradicate the negative regulatory activity.162 Mutation or loss of the PTEN/MMAC-1 gene product is almost exclusively limited to GBMs,122,184 and these aberrations appear to occur at the same frequencies whether EGFR is amplified or not.83 Additional tumor suppressor genes might therefore reside on chromosome 10 besides PTEN/MMAC-1.1,69,160,161 About 30 to 50% of gliomas also have a deletion of the extracellular domain of EGFR.101 This deletion results in a truncated receptor that is constitutively active in which signals are continually sent to the nucleus to initiate proliferation.
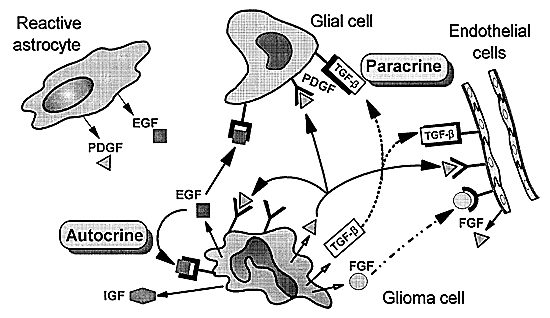
PDGF and its receptors differ from EGFR in that they have few mutations. Most GBMs, however, overexpress at least one PDGF chain and its respective receptor, and the most common form of overexpression is the PDGFA chain and PDGF-a receptor (Fig. 3).45,48 The PDGF-a receptor can bind all three isoforms, suggesting that an autocrine loop forms (Fig. 4) for this growth factor similar to the autocrine behavior of EGFR. Immunohistochemistry and in situ hybridization have clarified that the PDGFA chain and the PDGF-areceptor are preferentially expressed in tumor cells whereas the PDGFB chain and the PDGF-b receptor are highly expressed in proliferating endothelial cells within the tumor.51,53,115 Normal brain tissue also expresses the PDGF-b receptor and the PDGFA chain; however, the PDGF-b receptor will only bind the PDGFB chain so it is inactive in normal brain.91 Consequently, the preferential expression of the PDGF-b receptor in the endothelial component of gliomas may be related to the angiogenesis observed in these high-grade tumors.116
Other types of genes are also aberrant in GBMs and might contribute to their malignant and invasive phenotype. How these genes accomplish that task is undefined. For example, the DCC (deleted in colon cancer) gene is mapped to chromosome 18q21.31 The gene is a transmembrane cell adhesion molecule of the neural cell adhesion molecule (NCAM) family. As its name implies, DCC was discovered in colon cancer. High-grade gliomas exhibit abnormalities of chromosome 18, and several molecular studies have indicated that it is deleted in GBMs28,144 and in some Grade II tumors.144 Cell guidance molecules may therefore be involved in tumorigenesis.30 The novel gene D2-2 is overexpressed in fresh GBM tissue and cell lines and is highly expressed in recurrent tumor tissue.148 No known sequence homology has been determined for this gene yet it functions during fetal development where it is expressed about 28-fold higher than in normal adult brains.149
The list of genetic lesions associated with the different grades of gliomas continues to increase. Several investigators have suggested that multiple pathways lead to the formation of GBMs.181 They concluded that GBMs could be distinguished on the basis of genetic defects even though they are histopathologically indistinguishable from other gliomas.
One pathway is the progressive stepwise evolution from a low grade to the more malignant anaplastic astrocytoma and finally to the GBM. The mutations associated with this pathway would include allelic loss of 17p or TP53 mutations and PDGF activation, allelic loss on 22q (astrocytoma), followed by allelic loss of 13q, 9p and 19q (anaplastic astrocytoma) to the GBM (Fig. 1). Gliomas that exhibit these aberrant characteristics seldom have EGFR amplification or loss of chromosome 10.181 This progressive pathway creates what some investigators call secondary or type 1 GBMs.185 Patients diagnosed with this progressive tumor tend to be young (mean age, 39 years) and to have had a less malignant tumor at the time of their original diagnosis.
In contrast, the second pathway represents GBMs that arise de novo or very rapidly from a pre-existing tumor. EGFR amplification and allelic loss on chromosome 10 are the hallmarks of this pathway (Fig. 1).110,173 These tumors are usually associated with older patients (mean age, 55 years) with no history of a lower grade tumor. These tumors are considered primary or type 2 GBMs.185 In addition to EGFR and allelic loss on chromosome 10, the MDM2 gene product also appears to be restricted to the primary de novo pathway.126 A third pathway is defined by the absence of either 17p allelic loss or TP53 mutations or EGFR amplification.181
Although additional data are needed to elucidate these pathways, genetic markers may be able to distinguish these subsets. For example, data strongly support that GBMs that have progressed from lower grade tumors have a high percentage of TP53 mutations (67%) and a low frequency or absence of EGFR amplification.129,185 These pathways may also be associated with gender differences181,185 and may respond differently to specific therapies.193 Investigations are underway to determine if the genetic lesions identified in these subsets of gliomas have prognostic significance.21
Pilocytic Astrocytomas
Pilocytic astrocytomas tend to occur in children and young adults and show no clear sex preference. These tumors arise throughout the CNS43 but have a predilection for the cerebellum and region of the third ventricle, the optic nerves, and the thalamus. Juvenile pilocytic astrocytomas are more common than either protoplasmic or gemistocytic astrocytomas and are remarkable for maintaining their low-grade status for long periods. They rarely demonstrate progressive behavior.
Pilocytic tumors have not been analyzed in great detail because most of these tumors are biopsied. Of the 49 cases of pilocytic astrocytomas reported in the cytogenetic literature,153 all but five occurred in children under the age of 19 years. Of these 49 cases, 31 contained no detectable chromosome abnormalities. In the cases with abnormal chromosomes, the most common finding was the gain of chromosome 7 (5 of 44 cases). Two additional cases had a structural rearrangement of chromosome 7.
Molecular analyses of pilocytic tumors have focused on the TP53 gene.62,153 Of 125 tumors analyzed, 18 contained a loss of 17p. The loss of heterozygosity, however, was not within the TP53 gene. The most common loss appeared to be in the telomeric region (17p13.3) of chromosome 17194 or in the 17q arm,176 where the NF-1 gene (17q11.2) is located. The allelic loss detected on chromosome 17q is of interest because pilocytic astrocytomas are often associated with neurofibromatosis type 1.43 Based on these limited data, a gene or genes other than the TP53 gene appear to be the targeted genetic loss in pilocytic astrocytomas. Other molecular studies include analyses of chromosomes 1p,9 1q,9 9p,2,6,42 10,97 11p,120 12q,127 and 22q,186 but each study included only a small number of cases. Additional material must be analyzed before the role of specific genes associated with pilocytic astrocytomas can be determined.
Pleomorphic Xanthoastrocytomas
These tumors represent about 1% of the astrocytic neoplasms and typically develop in children and young adults without a sex bias.140 Cytogenetic studies performed on untreated tissue have reported numerical and structural abnormalities, but no consistent abnormality has been detected.153 Molecular studies of a small number of tumors have reported mutations in TP53, but no other allelic loss was identified.98,111
Giant Cell Glioblastomas
Also known as tuberous sclerosis, giant cell glioblastomas are a histological variant of GBMs. They account for less than 5% of the grade IV tumors, and their incidence peaks in the middle of the fifth decade.140 The giant cells within the tumor are the hallmark of this neoplasm. Two molecular investigations have identified TP53 mutations in more than 75% of the tumors analyzed.95,113 These same reports have also indicated that this group of tumors lacks other genetic alterations often associated with GBMs, such as EGFR, CDK4, and CDKN2 (MTS1/p16). The tuberous sclerosis complex (TSC) is an autosomal dominantly inherited disease characterized by the development of benign tumors and hamartomas. Among other problems, these patients also develop subependymal giant-cell astrocytomas.140 One study of astrocytomas and ependymomas has reported a reduced expression of tuberin, the protein product of the TSC complex. The protein product was completely absent in the one giant cell glioblastoma that has been analyzed.192
Ependymal Tumors
Ependymomas
Historically, the term ependymoblastoma referred to anaplastic ependymomas. Now, however, the term connotes embryonal tumors. Ependymomas develop from the ciliated epithelium that lines the ventricles of the brain and spinal canal. Embryologically, this cell layer is related to glia. When ependymal cells become transformed, they frequently express this glial heritage by acquiring a more astrocytic morphology. Ependymomas are most common during the first two decades of life,140 and their incidence is slightly higher for males than it is for females. They are more clearly defined from surrounding brain than other gliomas, although ependymomas demonstrate a spectrum of anaplasia. As with astrocytic tumors, histologic grade and survival do not appear to be correlated.
Summarizing the cytogenetic studies reviewed elsewhere,153 the most frequent abnormality is the loss of whole chromosome 22 in about 30% of the cases. Chromosome 22q is also involved in a number of structural rearrangements with most of the breakpoints localized to 22q12.153 The association between the loss of chromosome 22 and the location of the merlin gene at 22q12 is of interest. Patients with neurofibromatosis type 2 (NF-2) have a mutation in the merlin gene. They also have an increased incidence of intramedullary spinal ependymomas although intracranial ependymomas are rare.46 Abnormalities affecting the gain of chromosome 7 and the loss of 9p found so commonly in astrocytomas are not observed in ependymomas. Although far fewer ependymomas have been analyzed than astrocytomas, the former tend to lose chromosome 13q, the chromosome that contains the tumor suppressor gene, RB.62
Although a patient with a TP53 germ line mutation (Li-Fraumeni syndrome) has been reported to have developed intracranial ependymoma,94 molecular studies have rarely identified mutations of TP53.33,106,107,165 Another tumor suppressor gene may reside on 17p. One investigation reported that 50% of pediatric tumors had allelic loss on 17p, but the loss was distal to the TP53 gene. This allelic loss is similar to that identified in medulloblastomas.146 Genes associated with the cell cycle have also been found to be normal in ependymomas,142 and there is no evidence of EGFR amplification.11
This evidence confirms that the genetic lesions in astrocytomas are rare in ependymomas. These two types of brain tumors may therefore have different evolutionary pathways. Investigations of chromosome 22 suggest that a potential tumor suppressor gene resides there. A likely candidate gene is NF-2. One report detected mutations in the NF-2 gene,138 but other more extensive studies have failed to confirm this finding.11,54,138,158,182 Thus, more studies are needed to detect and clarify potential steps in the evolution of ependymomas.
Choroid Plexus Papillomas
This rare neoplasm most often occurs during the first decade of life and only rarely in adults. This tumor mimics its parental tissue by secreting cerebrospinal fluid and is one reason that hydrocephalus develops. The cells of the tumor appear strikingly normal and essentially free of mitotic activity. Only the crowding of cells identifies its neoplastic component. The number of cases reporting cytogenetic and molecular studies is limited. Half of the reported cases have normal karyotypes and the other half have numerical and structural abnormalities.153 Although choroid plexus papillomas have been identified in families with Li-Fraumeni syndrome,70 no other informative molecular studies have been performed.
Oligodendrogliomas and Mixed Tumors
Oligodendrogliomas
These tumors occur primarily in adults with a peak incidence in the fifth and sixth decades of life.140 Children can also develop oligodendrogliomas. The mean age of pediatric patients with supratentorial tumors is 10 years and 7.5 years for those with infratentorial tumors.164 Oligodendrogliomas account for 10% of the gliomas diagnosed and are usually considered slow growing.140The location of oligodendrogliomas is roughly related to the amount of white matter in the different lobes of the brain. Although these tumors arise in white matter, they tend to infiltrate the cerebral cortex more than astrocytomas of similar grade. Histologically, oligodendroglial tumors comprise a continuous spectrum that ranges from very well-differentiated neoplasms to malignant invasive tumors. Similar features (i.e., high cell density, mitotic activity and necrosis) are used to grade oligodendrogliomas. How these markers correlate with survival is controversial. With increasing anaplasia, oligodendrogliomas begin to appear more astrocytic, and they can develop areas of necrosis.
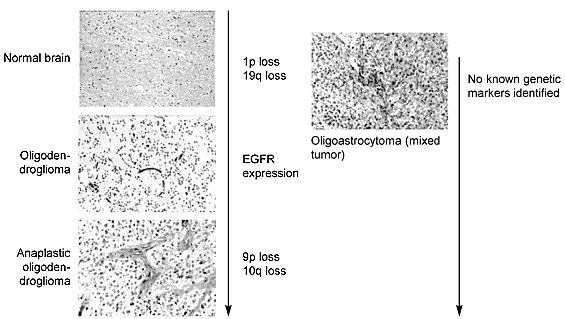
As primary untreated tumors, oligodendrogliomas often have normal G-banded karyotypes. The single sex chromosome is lost in about 25% of the cases, and chromosome 7 is gained in about 5% of the cases. Structural abnormalities are rare, although several have been localized to chromosome 1p and chromosome 22q. These findings are reviewed elsewhere.153
Molecular analyses have been informative in defining the genetic lesions associated with this tumor (Fig. 5). Allelic loss on chromosomes 1p and 19q120,121 appears to be preferential for oligodendrogliomas.7,128,178,197 The most frequent allelic loss, which occurs on chromosome 19q, has been observed in 50 to 80% of the tumors analyzed4,8,75,178 despite the lack of cytogenetic evidence of numerical or structural abnormalities of this chromosome.153
A putative tumor suppressor gene has been mapped to 19q13.2-q13.3.139,197 Many genes known to be lost or mutated in tumors reside on chromosome 19q, (e.g., DNA repair genes, ERCC1, ERCC2, XRCC, and DNA ligase, cell cycle gene, BAX, and TGF-b1).96A protein serine-threonine phosphatase is being evaluated as the potential tumor suppressor gene.198 Chromosome 1p allelic loss has been identified in 40% to 97% of the tumors. These disparate results frequently reflect different probes or methods of analysis.9,75,128Typically, tumors carrying 1p deletions also carry 19q allelic loss.8,75,128
The location of a potential tumor suppressor gene(s) is not well defined for chromosome 1p. Two potential sites have been localized: one at 1p35-p36 and a second site closer to the centromere.7 Other allelic losses have been reported for chromosomes 4q, 11p and 22q but await additional studies to determine their relative importance.128,187 Occasionally, oligodendrogliomas have mutations in the TP53 gene but far less frequently than observed in astrocytic tumors.88,107,129 Allelic loss or mutations of TP53 occur in less than 15% of the oligodendrogliomas.88,107,128,129 Immunoreactivity, however, has been identified in a much higher percentage,76,112 and these differences are yet to be resolved.
Although these tumors are considered slow growing, many develop anaplasia in the form of increased cellularity, nuclear atypia, cellular pleomorphism, and high levels of mitotic activity. The anaplasia can be accompanied by angiogenesis and the proliferation of vessels and necrosis. The genetic lesions associated with these histological changes are not well defined for anaplastic oligodendrogliomas. Nonrandom allelic loss on chromosomes 9p and 10q is a possibility.128,196 A potential target gene on 9p21 is the cell-cycle inhibitor, CDKN2A (MTS1/p16). However, one report found no allelic loss or mutations in the CDKN2A gene.142 There is also no evidence that the recently cloned PTEN/MMAC-1 tumor suppressor gene on 10q23 is the target of the 10q loss observed in oligodendrogliomas.80,162 Isolated reports suggest that one of several other genes is co-amplified80,162 with EGFR amplification.
Interpreting these studies and the genetic lesions identified in grade III oligodendrogliomas is difficult because most of these tumors have been treated with radiation, chemotherapy, or both. It is unclear whether the changes are a response to treatment or whether they were already present and selected as the resistant cell surviving treatment. Additional studies are needed to clarify these issues.
Mixed Tumors
Mixed tumors are composed of mixtures of oligodendroglial and astrocytic cells. The proportion of cells in this mixture varies considerably and is therefore a frequent point of contention among neuropathologists. The combination of glial cells most often observed in a mixed tumor is fibrillary astrocytes and oligodendrocytes. Mixtures of astrocytes and ependymal cells can occur but are thought to be rare. Such mixtures are also difficult to separate from ependymal tumors that have begun to acquire astrocytic phenotypes as discussed above. The cytogenetic literature has described 21 cases of mixed glioma, which are reviewed elsewhere.153 Summarized, however, the pattern of gain and loss in oligoastrocytomas is similar to that of oligodendrogliomas but without an astrocytic component. Again, more tumors must be analyzed to determine the presence of a nonrandom event.
Molecular studies have also been unable to identify a consistent genetic lesion that would indicate oligoastrocytomas are genetically distinct from either oligodendrogliomas or astrocytomas.128 A microdissection study of three different tumors determined that the different cellular components carried the same genetic lesions.75 About 30% of oligoastrocytomas carry genetic lesions often found in astrocytic gliomas, especially TP53 mutations and loss of heterozygosity on 17p.88,128 Furthermore, oligoastrocytomas with 17p loss or TP53 mutations show no allelic loss on 1p and 19q and vice versa. An extensive comparison of allelic loss in astrocytic tumors and oligodendrogliomas for chromosomes 1p, 17p and 19q suggested that mixed tumors have two genetic subsets—one genetically related to astrocytomas and the other to oligodendrogliomas.129
When mixed tumors acquire anaplastic features, they are also thought to acquire changes in 9p, 10, and 11p with occasional amplification of the EGFR gene or changes similar to the progressive changes of the anaplastic oligodendroglioma and astrocytoma.128 Again, more cases are needed for analysis before these patterns can be confirmed.
Brain Tumors from Mesenchymal Tissues
Gliosarcomas
Gliosarcomas are thought to originate when gliomas generate reactive responses that cause vascular cells, vascular adventitia, and fibroblasts of the meninges to undergo cell division. In most instances this proliferation is benign. Occasionally, however, these processes become transformed and create a neoplasm containing transformed cells of glial and mesenchymal origins.
Gliosarcomas are very difficult to analyze cytogenetically unless tumor cells can be cloned directly from a freshly resected tumor. Without cloning, neither morphology nor intermediate filament staining can determine if a clonal abnormality derives from the glial or sarcomatous components. However, once a clonal cytogenetic abnormality has been established, fluorescent in situ hybridization could be used with paraffin-embedded tissue from the same patient to determine with which component the abnormality was associated.
Of six cytogenetic analyses of gliosarcomas in patients between 33 and 80 years old, four cases gained chromosome 7 while three cases lost chromosome 10. Structural abnormalities were common, but chromosome 9 was involved most frequently (five cases). Three cases had a translocation involving chromosome 9, but the breakpoints were different in each case. Molecular analyses of fresh tumor tissue are notably absent although occasional allelic loss and mutations of the TP53 gene have been detected.35,78
Meningiomas
No single meningeal layer has been identified as the tumor-forming component of meningiomas. The incidence of these tumors is about 15% of all primary brain tumors.140 They can occur almost anywhere in the brain, but their greatest incidence is in the cerebral convexities and their lowest is in the pineal region. Although the overall incidence of spinal cord meningiomas is low, they still constitute the largest group of tumors in this region. Meningiomas develop in the middle decades of life and in the elderly, primarily in females (especially spinal meningiomas). They are rare in children, accounting for less than 2% of intracranial tumors.3 A familial occurrence, mostly associated with von Recklinghausens’s disease, is known. Most cases, however, are sporadic.
The current grading system recognizes three grades of meningiomas: benign (Grade I), atypical (Grade II), and anaplastic (Grade III). Benign tumors represent about 94% of the meningiomas; 5% are atypical and 1% are anaplastic.15,140 Atypical and anaplastic meningiomas pose the greatest risk of recurrence and therefore prognostic markers for those at risk would be helpful in determining the appropriate treatment for patients, especially children. Meningiomas have a wide range of histopathological appearances. The most common subtypes are meningothelial, fibrous, and transitional.140
Meningiomas were among the first solid tumors in which a nonrandom loss of a whole chromosome was identified.200 Monosomy 22 is the most consistent chromosomal aberration along with the deletion of chromosome 22.199 Despite its prevalence, this abnormality is independent of the tumor’s biological behavior. Tumor progression is thought to be more closely associated with the rearrangement or loss of chromosomes 1p, 14q, or both. The cytogenetic analysis of meningiomas is reviewed elsewhere.153 In general, molecular studies have reflected the chromosome abnormalities, especially for chromosome 22.25,26,82,92,141
Neurofibromatosis patients develop multiple meningiomas,137 and the NF-2 gene is localized to chromosome 22q12.136,166 Allelic loss on chromosome 22q and mutations in the NF-2 gene are observed in about 50 to 60% of the sporadic meningiomas25,26,141,150 and in meningiomas from patients with NF-2.150 This finding supports the hypothesis that the NF-2 locus encodes a tumor suppressor gene involved with meningiomas. In some studies mutations in NF-2 were as high as 70 to 80% when only fibroblastic and transitional meningiomas were included.189 In contrast, only 25% of the meningothelial meningiomas cases have contained an NF-2 mutation although this finding is controversial.47
Despite the association of meningiomas with NF-2 mutations, other sporadic patients and families with multiple meningiomas (familial) do not have a germ-line mutation in the NF-2 gene.117 Another locus on 22q may therefore encode a tumor suppressor gene. Allelic loss has been identified between 22q12.3 and the telomere, a region not associated with the NF-2 gene.133 Candidate genes under consideration are the
b-adaptin gene114 and MN1.79
Molecular studies that have detected allelic losses on chromosomes 1p5 and 14q93,157,167 support the cytogenetic data. However, additional allelic losses were detected for chromosomes 9q, 10q,82,131,157 and 17p.118,183 Each of these allelic losses occurs in tumors with more aggressive or invasive phenotypes.
Another phenotype beginning to emerge from the study of meningiomas is the aberrant expression of growth factors. In particular, the B chain of PDGF and the PDGF-b receptor have been identified.13,77,89,90 Their expression is suggestive of an autocrine mechanism. An immunohistochemical study further supports the possibility of an autocrine loop. Fresh meningioma tissue samples expressed the PDGF-B chain and PDGF-b receptor aberrantly, and this expression was localized to tumor tissue rather than to normal stroma.152Other growth factors identified in meningiomas include IGF-144,105,168 and TGF-b64,103 although their role in tumor progression awaits clarification.
Intracranial Schwann Cell Neoplasms
Schwannomas (neurilemmomas) are slow-growing, benign tumors that rarely undergo malignant changes. The most common Schwann cell neoplasm occurs on the eighth cranial nerve and less frequently on the fifth cranial nerve. Acoustic neuromas are derived from Schwann cells that envelop the vestibular branch of the 8th nerve. Most tumors are sporadic, but bilateral tumors are an indication of familial cases.140 Multiple peripheral schwannomas in the absence of neurofibromatosis features are characteristic of the newly described syndrome called schwannomatosis.87 The greatest incidence of this tumor is between the third and sixth decades of life with no sex predilection.
The NF-2 gene is considered the tumor suppressor gene responsible for the development of this neoplasm.57,58,151 The protein product of the NF-2 gene is called schwannomin/merlin, which is a member of a superfamily of proteins thought to play crucial roles in linking cell membrane proteins with the cytoskeleton.166 Cytogenetic20,132 and molecular studies have demonstrated that 40 to 60% of sporadic acoustic neurinomas have allelic loss on chromosome 22q.57,58,136,151,166 Immunohistochemical studies demonstrate the complete loss of schwannomin/merlin expression in schwannomas, further substantiating that eliminating the expression of schwannomin/merlin is an essential step in tumorigenesis.
Conclusion
At present no single chromosome abnormality clearly defines any one type or grade of brain tumor. The most common abnormality (the overrepresentation of chromosome 7) is present in about 60% of astrocytic tumors. It is unclear, however, if tumors lacking this abnormality represent a different subset of astrocytomas or whether it reflects a genetic instability that has by chance sorted human chromosomes in a way that provides a selective survival advantage to cells. When the changes in DNA have been assessed, patterns of change have been observed. Clearly, a series of step-wise activations and loss of genes occur during tumor progression, and these changes appear to be different types of brain tumors. Less clear is how tumors are initiated. Genetic changes in key genes such as TP53 are known to be early changes. Not all tumors, however, have an abnormality of this gene, suggesting that alternative pathways or mechanisms remain to be defined.
As new genetic abnormalities are defined, each has the potential to become a therapeutic target. Certainly, new drugs and gene therapy have expanded as such abnormalities have been defined. These new strategies differ from those of the past by specifically targeting tumor cells while sparing normal brain tissue.
Genetic lesions also offer the potential to be used as markers for diagnosis and prognosis, especially in genetic cancers like neuroblastomas. Such advances are in their infancy. This technology, however, has tremendous potential and will be part of the medical armament in the near future.
References
1. Albarosa R, Colombo BM, Roz L, et al: Deletion mapping of gliomas suggests the presence of two small regions for candidate tumor-suppressor genes in a 17-cM interval on chromosome 10q. Am J Hum Genet 58:1260-1267, 1996
2. Barker FG, Chen P, Furman F, et al: p16 deletion and mutation analysis in human brain tumors. J Neurooncol 31:17-23, 1997
3. Baumgartner JE, Sorenson JM: Meningioma in the pediatric population. J Neurooncol 29:223-228, 1996
4. Bello MJ, de Campos JM, Kusak ME, et al: Molecular analysis of genomic abnormalities in human gliomas. Cancer Genet Cytogenet 73:122-129, 1994
5. Bello MJ, de Campos JM, Kusak ME, et al: Allelic loss at 1p is associated with tumor progression of meningiomas. Genes Chromosomes Cancer 9:296-298, 1994
6. Bello MJ, de Campos JM, Vaquero J, et al: Molecular and cytogenetic analysis of chromosome 9 deletions in 75 malignant gliomas.Genes Chromosomes Cancer 9:33-41, 1994
7. Bello MJ, Leone PE, Nebreda P, et al: Allelic status of chromosome 1 in neoplasms of the nervous system. Cancer Genet Cytogenet 83:160-164, 1995
8. Bello MJ, Leone PE, Vaquero J, et al: Allelic loss at 1p and 19q frequently occurs in association and may represent early oncogenic events in oligodendroglial tumors. Int J Cancer 64:207-210, 1995
9. Bello MJ, Vaquero J, de Campos JM, et al: Molecular analysis of chromosome 1 abnormalities in human gliomas reveals frequent loss of 1p in oligodendroglial tumors. Int J Cancer 57:172-175, 1994
10. Bigner SH, Mark J, Mahaley MS, et al: Patterns of the early, gross chromosomal changes in malignant human gliomas. Hereditas 101:103-113, 1984
11. Bijlsma EK, Voesten AMJ, Bijleveld EH, et al: Molecular analysis of genetic changes in ependymomas. Genes Chromosomes Cancer 13:272-277, 1995
12. Black P, Westphal M (eds.): Growth factors in brain tumors. J Neurooncol 353(3):193-372, 1997
13. Black PM, Carroll R, Glowacka D, et al: Platelet-derived growth factor expression and stimulation in human meningiomas. J Neurosurg 81:388-393, 1994
14. Brodeur GM, Fong CT: Molecular biology and genetics of neuroblastoma. Cancer Genet Cytogenet 41:153-174, 1989
15. Burger PC, Scheithauer BW, Vogel FS: Brain Tumors, in Surgical Pathology of the Nervous System and its Coverings. New York: Churchill Livingstone, 1991, pp 193-437
16. Cavenee WK, Dryja TP, Phillips RA: Expression of recessive alleles by chromosomal mechanisms in retinoblastomas. Nature 305:779-784, 1983
17. Chattopadhyay P, Rathore A, Mathur M, et al: Loss of heterozygosity of a locus on 17p13.3 independent of p53, is associated with higher grades of astrocytic tumours. Oncogene 15:871-874, 1997
18. Collins VP, James CD: Gene and chromosomal alterations associated with the development of human gliomas. FASEB J 7:926-930, 1993
19. Coons SW, Johnson PC, Shapiro JR: Cytogenetic and flow cytometry DNA analysis of regional heterogeneity in a low grade glioma. Cancer Res 55:1569-1577, 1995
20. Couturier J, Delattre O, Kujas M, et al: Assessment of chromosome 22 anomalies in neurinomas by combined karyotype and RFLP analyses. Cancer Genet Cytogenet 45:55-62, 1990
21. Dalrymple SJ, Herath JF, Ritland SR, et al: Use of fluorescence in situ hybridization to detect loss of chromosome 10 in astrocytomas. J Neurosurg 83:316-323, 1995
22. Daumas-Duport C: Histological grading of gliomas. Current Opinion Neurol Neurosurg 5:924-931, 1992
23. de Fromentel CC, Soussi T: TP53 tumor suppressor gene: A model for investigating human mutagenesis. Genes Chromosomes Cancer 4:1-15, 1992
24. Downward J, Yarden Y, Mayes E, et al: Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature 307:521-527, 1984
25. Dumanski JP, Carlbom E, Collins VP, et al: Deletion mapping of a locus on human chromosome 22 involved in the oncogenesis of meningioma. Proc Natl Acad Sci USA 84:9275-9279, 1987
26. Dumanski JP, Rouleau GA, Nordenskjold M, et al: Molecular genetic analysis of chromosome 22 in 81 cases of meningioma.Cancer Res 50:5863-5867, 1990
27. Ekstrand AJ, Sugawa N, James CD, et al: Amplified and rearranged EGFR genes in human glioblastomas reveal deletions of sequences encoding portions of the N-and/or C-terminal tails. Proc Natl Acad Sci USA 89:4309-4313, 1992
28. Ekstrand BC, Bigner SH, Fearon ER: DCC gene mutations and loss of expression in glioblastomas. Proc Am Assoc Cancer Res 36:574, 1995 (Abstract)
29. El-Azouzi M, Chung RY, Farmer GE, et al: Loss of distinct regions on the short arm of chromosome 17 associated with tumorigenesis of human astrocytomas. Proc Natl Acad Sci USA 86:7186-7190, 1989
30. Fearon ER: DCC: Is there a connection between tumorigenesis and cell guidance molecules? Biochem Biophys Acta Rev Cancer 1288:M17-M23, 1996
31. Fearon ER, Cho KR, Nigro JM, et al: Identification of a chromosome 18q gene that is altered in colorectal cancers. Science 247:49-56, 1990
32. Fearon ER, Vogelstein B: A genetic model for colorectal tumorigenesis. Cell 61:759-767, 1990
33. Fink KL, Rushing EJ, Schold SC, et al: Infrequency of p53 gene mutations in ependymomas. J Neurooncol 27:111-115, 1996
34. Fleming TP, Saxena A, Clark WC, et al: Amplification and/or overexpression of platelet-derived growth factor receptors and epidermal growth factor receptor in human glial tumors. Cancer Res 52:4550-4553, 1992
35. Frankel RH, Bayona W, Koslow M, et al: p53 mutations in human malignant gliomas: Comparison of loss of heterozygosity with mutation frequency. Cancer Res 52:1427-1433, 1992
36. Fults D, Brockmeyer D, Tullous MW, et al: p53 mutation and loss of heterozygosity on chromosome 17 and 10 during human astrocytoma progression. Cancer Res 52:674-679, 1992
37. Fults D, Pedone C: Deletion mapping of the long arm of chromosome 10 in glioblastoma multiforme. Genes Chromosomes Cancer 7:173-177, 1993
38. Fults D, Pedone CA, Thomas GA, et al: Allelotype of human malignant astrocytoma. Cancer Res 50:5784-5789, 1990
39. Fults D, Petronio J, Noblett BD, et al: Chromosome 11p15 deletions in human malignant astrocytomas and primitive neuroectodermal tumors. Genomics 14:799-801, 1992
40. Fults D, Tippets RH, Thomas GA, et al: Loss of heterozygosity for loci on chromosome 17p in human malignant astrocytoma.Cancer Res 49:6572-6577, 1989
41. Furnari FB, Huang HJS, Cavenee WK: Genetics and malignant progression of human brain tumors, in Ponder BAJ, Cavenee WK, Solomon E (eds): Genetics and Cancer: A Second Look. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 1995, pp 233-275
42. Gao L, Liu L, van Meyel D, et al: Lack of germ-line mutations of CDK4, p16INK4A, and p15INK4B in families with glioma. Clinical Cancer Res 3:977-981, 1997
43. Giannini C, Scheithauer BW: Classification and grading of low-grade astrocytic tumors in children. Brain Pathol 7:785-798, 1997
44. Glick RP, Unterman TG, Woude MV, et al: Insulin and insulin-like growth factors in central nervous system tumors. Part V: Production of insulin-like growth factors I and II in vitro. J Neurosurg 77:445-450, 1992
45. Guha A, Dashner K, Black PM, et al: Expression of PDGF and PDGF receptors in human astrocytoma operation specimens supports the existence of an autocrine loop. Int J Cancer 60:168-173, 1995
46. Hamilton RL, Pollack IF: The molecular biology of ependymomas. Brain Pathol 7(2):807-822, 1997
47. Harada T, Irving RM, Xuereb JH, et al: Molecular genetic investigation of the neurofibromatosis type 2 tumor suppressor gene in sporadic meningioma. J Neurosurg 84:847-851, 1996
48. Harsh GR, Keating MT, Escobedo JA, et al: Platelet derived growth factor (PDGF) autocrine components in human tumor cell lines.J Neurooncol 1:1-12, 1990
49. Hashimoto N, Ichikawa D, Arakawa Y, et al: Frequent deletions of material from chromosome arm 1p in ogliodendroglial tumors revealed by double-target fluorescence in situ hybridization and microsatellite analysis. Genes Chromosomes Cancer 14:295-300, 1995
50. He J, Olson JJ, James CD: Lack of p16INK4 or retinoblastoma protein (pRb), or amplification-associated overexpression of cdk4 is observed in distinct subsets of malignant glial tumors and cell lines. Cancer Res 55:4833-4836, 1995
51. Hermanson M, Funa K, Hartman M, et al: Platelet-derived growth factor and its receptors in human glioma tissue: Expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res 52:3213-3219, 1992
52. Hermanson M, Funa K, Koopmann J, et al: Association of loss of heterozygosity on chromosome 17p with high platelet-derived growth factor receptor expression in human malignant gliomas. Cancer Res 56:164-171, 1996
53. Hermanson M, Nistér M, Betsholtz C, et al: Endothelial cell hyperplasia in human glioblastoma: Coexpression of mRNA for platelet-derived growth factor (PDGF) B chain and PDGF receptor suggests autocrine growth stimulation. Proc Natl Acad Sci USA 85:7748-7752, 1988
54. Hoang-Xuan K, Merel P, Vega F, et al: Analysis of the NF2 tumor-suppressor gene and of chromosome 22 deletions in gliomas. Int J Cancer 60:478-481, 1995
55. Hollstein M, Sidransky D, Vogelstein B, et al: p53 mutations in human cancers. Science 253:49-53, 1991
56. Israel MA: Molecular biology of childhood neoplasms, in Mendelsohn J, Howley PM, Israel MA, et al (eds): The Molecular Basis of Cancer. W.B. Saunders: Philadelphia, 1995, pp 294-316
57. Jacoby LB, MacCollin M, Barone R, et al: Frequency and distribution of NF2 mutations in schwannomas. Genes Chromosomes Cancer 17:45-55, 1996
58. Jacoby LB, MacCollin M, Louis DN, et al: Exon scanning for mutation of the NF2 gene in schwannomas. Human Mol Genet 3:413-419, 1994
59. James CD, Carlbom E, Dumanski JP, et al: Clonal genomic alterations in glioma malignancy stages. Cancer Res 48:5546-5551, 1988
60. James CD, Carlbom E, Nordenskjold M, et al: Mitotic recombination of chromosome 17 in astrocytomas. Proc Natl Acad Sci USA 86:2858-2862, 1989
61. James CD, Collins VP: Molecular genetic characterization of CNS tumor oncogenesis. Adv Cancer Res 58:121-142, 1992
62. James CD, He J, Carlbom E, et al: Loss of genetic information in central nervous system tumors common to children and young adults. Genes Chromosomes Cancer 2:94-102, 1990
63. James CD, He J, Collins VP, et al: Localization of chromosomes 9p homozygous deletions in glioma cell lines with markers constituting a continuous linkage group. Cancer Res 53:3674-3676, 1993
64. Johnson MD, Federspiel CF, Gold LI, et al: Transforming growth factor-b and transforming growth factor-b receptor expression in human meningioma cells. Am J Pathol 141:633-642, 1992
65. Jost CA, Marin MC, Kaelin WG, Jr: p73 is a human p53-related protein that can induce apoptosis. Nature 389:191-194, 199
66. Kaghad M, Bonnet H, Yang A, et al: Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 90:809-819, 1997
67. Karlbom AE, James CD, Boethius J, et al: Loss of heterozygosity in malignant gliomas involves at least three distinct regions on chromosome 10. Hum Genet 92:169-174, 1993
68. Kieser A, Weich HA, Brandner G, et al: Mutant p53 potentiates protein kinase C induction of vascular endothelial growth factor expression. Oncogene 9:963-969, 1994
69. Kimmelman AC, Ross DA, Liang BC: Loss of heterozygosity of chromosome 10p in human gliomas. Genomics 34:250-254, 1996
70. Kleihues P, Schauble B, zur Hausen A, et al: Tumours associated with p53 germline mutations. A synopsis of 91 families. Am J Pathol 150:1-13, 1997
71. Kleihues P, Soylemezoglu F, Schäuble B, et al: Histopathology, classification, and grading of gliomas. Glia 15:211-221, 1995
72. Koga H, Zhang S, Kumanishi T, et al: Analysis of p53 gene mutations in low- and high-grade astrocytomas by polymerase chain reaction-assisted single-strand conformation polymorphism and immunohistochemistry. Acta Neuropathol (Berl) 87:225-232, 1994
73. Kok K, Naylor SL, Buys CHCM: Deletion of the short arm of chromosome 3 in solid tumors and the search for suppressor genes.Adv Cancer Res 27-91, 1997
74. Kraus JA, Bolln C, Wolf HK, et al: TP53 Alterations and clinical outcome in low grade astrocytomas. Genes Chromosomes Cancer 10:143-149, 1994
75. Kraus JA, Koopmann J, Kaskel P, et al: Shared allelic losses on chromosomes 1p and 19q suggest a common origin of oligodendroglioma and oligoastrocytoma. J Neuropathol Exp Neurol 54:91-95, 1995
76. Kros JM, Godschalk JJ, Krishnadath KK, et al: Expression of p53 in oligodendrogliomas. J Pathol 171:285-290, 1993
77. Kuratsu JI, Seto H, Kochi M, et al: Expression of PDGF, PDGF-receptor, EGF-receptor and sex hormone receptors on meningioma. Acta Neurochir 131:289-293, 1994
78. Kyritsis AP, Bondy ML, Xiao M, et al: Germline p53 gene mutations in subsets of glioma patients. J Natl Cancer Inst 86:344-349, 1994
79. Lekanne Deprez RH, Riegman PH, Groen NA, et al: Cloning and characterization of MN1, a gene from chromosome 22q11, which is disrupted by a balanced translocation in a meningioma. Oncogene 10:1521-1528, 1995
80. Li J, Yen C, Liaw D, et al: PTEN, a putative protein tyrosine phospatase gene mutated in human brain, breast, and prostate cancer.Science 275:1943-1947, 1997
81. Libermann TA, Nusbaum HR, Razon N, et al: Amplification and overexpression of the EGF receptor gene in primary human glioblastomas. J Cell Sci 3:161-172, 1985
82. Lindblom A, Ruttledge M, Collins VP, et al: Chromosome deletions in anaplastic meningiomas suggest multiple regions outside chromosome 22 as important in tumor progression. Int J Cancer 56:354-357, 1994
83. Liu W, James CD, Frederick L, et al: PTEN/MMAC1 mutations and EGFR amplification in glioblastomas. Cancer Res 57:5254-5257, 1997
84. Louis DN: A molecular genetic model of astrocytoma histopathology. Brain Pathol 7:755-764, 1997
85. Louis DN: The p53 gene and protein in human brain tumors. J Neuropathol Exper Neurol 53:11-21, 1994
86. Louis DN, von Deimling A, Chung RY, et al: Comparative study of p53 gene and protein alterations in human astrocytic tumors. J Neuropathol Exper Neurol 52(1):31-38, 1993
87. MacCollin M, Woodfin W, Kronn D, et al: Schwannomatosis: A clinical and pathologic study. Neurology 46:1072-1079, 1996
88. Maintz D, Fiedler K, Koopmann J, et al: Molecular genetic evidence for subtypes of oligoastrocytomas. J Neuropathol Exp Neurol 56:1098-1104, 1997
89. Mauro A, Di Sapio A, Mocellini C, et al: Control of meningioma cell growth by platelet-derived growth factor (PDGF). J Neurol Sci 131:135-143, 1995
90. Maxwell M, Galanopoulos T, Hedley-Whyte ET, et al: Human meningiomas co-express platelet-derived growth factor (PDGF) and PDGF-receptor genes and their protein products. Int J Cancer 46:16-21, 1990
91. Maxwell M, Naber SP, Wolfe HJ, et al: Coexpression of platelet-derived growth factor (PDGF) and PDGF-receptor genes by primary human astrocytomas may contribute to their development and maintenance. J Clin Invest 86:131-140, 1990
92. Meese E, Blin N, Zang KD: Loss of heterozygosity and the origin of meningioma. Hum Genet 77:349-351, 1987
93. Menon AG, Rutter JL, von Sattel JP, et al: Frequent loss of chromosome 14 in atypical and malignant meningioma: Identification of a putative ‘tumor progression’ locus. Oncogene 14:611-616, 1997
94. Metzger AK, Sheffield VC, Duyk G, et al: Identification of a germ-line mutation in the p53 gene in a patient with an intracranial ependymoma. Proc Natl Acad Sci USA 88:7825-7829, 1991
95. Meyer-Puttlitz B, Hayashi Y, Waha A, et al: Molecular genetic analysis of giant cell glioblastomas. Am J Pathol 151:853-857, 1997
96. Mohrenweiser H, Olsen A, Archibald A, et al: Report of the third international workshop on human chromosome 19 mapping 1996.Cytogenet Cell Genet 74:161-186, 1996
97. Morse RP, Darras BT, Ye Z, et al: Clonal analysis of human astrocytomas. J Neurooncol 21:151-157, 1994
98. Munoz EL, Eberhard DA, Lopes MBS, et al: Proliferative activity and p53 mutation as prognostic indicators in pleomorphic xanthoastrocytoma. J Neuropathol Exp Neurol 55:606-609, 1996
99. Ng H, Lau K, Tse JYM, et al: Combined molecular genetic studies of chromosome 22q and the neurofibromatosis type 2 gene in central nervous system tumors. Neurosurgery 37:764-773, 1995
100. Nigro JM, Baker SJ, Preisinger AC, et al: Mutations in the p53 gene occur in diverse human tumour types. Nature 342:705-708, 1989
101. Nishikawa R, Ji XD, Harmon RC, et al: A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci USA 91:7727-7731, 1994
102. Nistér M, Claesson-Welsh L, Eriksson A, et al: Differential expression of platelet-derived growth factor receptors in human malignant glioma cell lines. J Biol Chem 266:16755-16763, 1991
103. Nitta T, Sato K, Okumura K: Transforming growth factor (TGF)-b like activity of intracranial meningioma and its effect on cell growth. J Neurol Sci 101:19-23, 1991
104. Nobori T, Miura K, Wu DJ, et al: Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature 368:753-756, 1994
105. Nordqvist, Peyrard M, Pettersson H, et al: A high ratio of insulin-like growth factor II/insulin-like growth factor binding protein 2 messenger RNA as a marker for anaplasia in meningiomas. Cancer Res 57:2611-2614, 1997
106. Ohgaki H, Eibl RH, Schwab M, et al: Mutations of the p53 tumor suppressor gene in neoplasms of the human nervous system.Mol Carcinog 8:74-80, 1993
107. Ohgaki H, Eibl RH, Wiestler OD, et al: p53 mutations in nonastrocytic human brain tumors. Cancer Res 51:6202-6205, 1991
108. Olopade OI, Jenkins RB, Ransom DT, et al: Molecular analysis of deletions of the short arm of chromosome 9 in human gliomas.Cancer Res 52:2523-2529, 1992
109. Ono Y, Tamiya T, Ichiakawa T, et al: Malignant astrocytomas with homozygous CDKN2/p16 gene deletions have higher Ki-67 proliferation indices. J Neuropathol Exp Neurol 55:1026-1031, 1996
110. Patt S, Thiel G, Maas S, et al: Chromosomal changes and correspondingly altered proto-oncogene expression in human gliomas. Value of combined cytogenetic and molecular genetic analysis. Anticancer Res 13:113-118, 1993
111. Paulus W, Lisle DK, Tonn JC, et al: Molecular genetic alterations in pleomorphic xanthoastrocytoma. Acta Neuropathol 91:293-297, 1996
112. Pavelic A, Hlavka V, Poljak M, et al: p53 Immunoreactivity in oligodendrogliomas. J Neurooncol 22:1-6, 1994
113. Peraud A, Watanabe K, Plate KH, et al: p53 mutations versus EGF receptor expression in giant cell glioblastomas. J Neuropathol Exp Neurol 56:1236-1241, 1997
114. Peyrard M, Fransson I, Xie Y-G, et al: Characterization of a new member of the human b-adaptin gene family from chromosome 22q12, a candidate meningioma gene. Hum Mol Genet 3:1393-1399, 1994
115. Plare KH, Breier G, Farrell CL, et al: Platelet-derived growth factor receptor-beta is induced during tumor development and upregulated during tumor progression in endothelial cells in human gliomas. Lab Invest 67:529-534, 1992
116. Plate KH, Breier G, Risau W: Molecular mechanisms of developmental and tumor angiogenesis. Brain Pathology 4:207-218, 1994
117. Pulst S-M, Rouleau GA, Marineau C, et al: Familial meningioma is not allelic to neurofibromatosis 2. Neurology 43:2096-2098, 1993
118. Pykett MJ, Landers J, George DL: Expression patterns of the p53 tumor suppressor gene and the mdm2 proto-oncogene in human meningiomas. J Neurooncol 32:39-44, 1997
119. Raffel C, Thomas G, Tishler DM, et al: Absence of p53 mutations in childhood central nervous system primitive neuroectodermal tumors. Neurosurgery 33:301-306, 1993
120. Ransom DT, Ritland SR, Kimmel DW, et al: Cytogenetic and loss of heterozygosity studies in ependymomas, pilocytic astrocytomas and oligodendrogliomas. Genes Chromosomes Cancer 5:348-356, 1992
121. Ransom DT, Ritland SR, Moertel CA, et al: Correlation of cytogenetic analysis and loss of heterozygosity studies in human diffuse astrocytomas and mixed oligo-astrocytomas. Genes Chromosomes Cancer 5:357-374, 1992
122. Rasheed BK, Stenzel TT, McLendon RE, et al: PTEN gene mutations are seen in high-grade but not in low-grade gliomas.Cancer Res 57:4187-4190, 1997
123. Rasheed BKA, Fuller GN, Friedman AH, et al: Loss of heterozygosity for 10q loci in human gliomas. Genes Chromosomes Cancer 5:75-82, 1992
124. Rasheed BKA, McLendon RE, Herndon JE, et al: Alterations of the TP53 gene in human gliomas. Cancer Res 54:1324-1330, 1994
125. Reifenberger G, Ichimura K, Reifenberger J, et al: Refined mapping of 12q13-q15 amplicons in malignant gliomas suggests CDK4/SAS and MDM2 as independent amplification targets. Cancer Res 56:5141-5145, 1996
126. Reifenberger G, Liu L, Ichimura K, et al: Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res 53:2736-2739, 1993
127. Reifenberger G, Reifenberger J, Ichimura K, et al: Amplification at 12q13-14 in human malignant gliomas is frequently accompanied by loss of heterozygosity at loci proximal and distal to the amplification site. Cancer Res 55:731-734, 1995
128. Reifenberger J, Reifenberger G, Liu L, et al: Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol 145:1175-1190, 1994
129. Reifenberger J, Ring GU, Gies U, et al: Analysis of p53 mutation and epidermal growth factor receptor amplification in recurrent gliomas with malignant progression. J Neuropathol Exp Neurol 55:822-831, 1996
130. Reisman D, Elkind NB, Roy B, et al: C-Myc enhances expression from the promoter of the p53 tumor suppressor gene through site-specific binding to downstream CACGTG motif. Cell Growth Differ 4:57-62, 1993
131. Rempel SA, Schwechheimer K, Davis RL, et al: Loss of heterozygosity for loci on chromosome 10 is associated with morphologically malignant meningioma progression. Cancer Res 53:2386-2392, 1993
132. Rey JA, Bello MJ, de Campos JM, et al: Cytogenetic analysis in human neurinomas. Cancer Genet Cytogenet 28:187-188, 1987
133. Rey JA, Bello MJ, de Campos JM, et al: Abnormalities of chromosome 22 in human brain tumors determined by combined cytogenetic and molecular genetic approaches. Cancer Genet Cytogenet 66:1-10, 1993
134. Rey JA, Bello MJ, Jimenez-Lara AM, et al: Loss of heterozygosity for distal markers on 22q in human gliomas. Int J Cancer 51:703-706, 1992
135. Ritland SR, Ganju V, Jenkins RB: Region-specific loss of heterozygosity on chromosome 19 is related to the morphologic type of human glioma. Genes Chromosomes Cancer 12:277-282, 1995
136. Rouleau GA, Mere P, Lutchman M, et al: Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature 363:515-521, 1993
137. Rouleau GA, Wertelecki W, Haines JL, et al: Genetic linkage of bilateral acoustic neurofibromatosis to a DNA marker on chromosome 22. Nature 329:246-248, 1987
138. Rubio M-P, Correa KM, Ramesh V, et al: Analysis of the neurofibromatosis 2 gene in human ependymomas and astrocytomas.Cancer Res 54:45-47, 1994
139. Rubio M-P, Correa KM, Ueki K, et al: The putative glioma tumor suppressor gene on chromosome 19q maps between APOC2 and HRC. Cancer Res 54:4760-4763, 1994
140. Russell DS, Rubinstein LJ: Pathology of Tumors of the Nervous System. Baltimore: Williams and Wilkins, 1989
141. Ruttledge MH, Xie Y-G, Han F-Y, et al: Deletions on chromosome 22 in sporadic meningioma. Genes Chromosomes Cancer 10:122-130, 1994
142. Sato K, Schauble B, Kleihues P, et al: Infrequent alterations of the p15, p16, CDK4 and cyclin D1 genes in non-astrocytic human brain tumours. Int J Cancer 66:305-308, 1996
143. Saxena A, Clark WC, Robertson JT, et al: Evidence for the involvement of a potential second tumor suppressor gene on chromosome 17 distinct from p53 in malignant astrocytomas. Cancer Res 52:6716-6721, 1992
144. Scheck AC, Coons SW: Expression of the tumor suppressor gene DCC in human gliomas. Cancer Res 53:5605-5609, 1993
145. Scheck AC, Mehta BM, Beikman MK, et al: BCNU-resistant human glioma cells with over-representation of chromosomes 7 and 22 demonstrate increased copy number and expression of platelet-derived growth factor genes. Genes Chromosomes Cancer 8:137-148, 1993
146. Scheurlen WG, Seranski P, Mincheva A, et al: High-resolution deletion mapping of chromosome arm 17p in childhood primitive neuroectodermal tumors reveals a common chromosomal disruption within the Smith-Magenis region, an unstable region in chromosome band 17p11.2. Genes Chromosomes Cancer 18:50-58, 1997
147. Schofield DE: Diagnostic histopathology, cytogenetics, and molecular markers of pediatric brain tumors. Neurosurg Clin N Am 3:723-738, 1992
148. Sehgal A: Molecular changes during the genesis of human gliomas. Semin Surg Oncol 14:3-12, 1998
149. Sehgal A, Keener C, Boynton AL, et al: Characterization of C4-2 as a tumor-suppressor gene in human brain tumors. J Surg Oncol 64:102-108, 1997
150. Seizinger BR, De la Monte SM, Atkins L: Molecular genetic approach to human meningioma: Loss of genes on chromosome 22.Proc Natl Acad Sci USA 84:5419-5423, 1987
151. Seizinger BR, Martuza RL, Gusella JF: Loss of genes on chromosome 22 in human acoustic neuroma. Nature 322:644-647, 1986
152. Shamah SM, Alberta JA, Giannobile WV, et al: Detection of activated platelet-derived growth factor receptors in human meningioma. Cancer Res 57:4141-4147, 1997
153. Shapiro JR, Scheck AC: Brain tumors, in Wolman SR, Sell S (eds): Cytogenetic Markers of Human Cancer. New Jersey: The Humana Press, Inc., 1997, pp 319-368
154. Shapiro WR: Therapy of adult malignant brain tumors: What have the clinical trials taught us? Semin Oncol 13:38-45, 1986
155. Shapiro WR, Shapiro JR, Walker RW: Central Nervous System, in Abeloff MD, Armitage JO, Lichter AS, et al (eds): Clinical Oncology. New York: Churchill Livingstone, 1995, pp 851-912
156. Sidransky D, Mikkelsen T, Schwechheimer K, et al: Clonal expansion of p53 mutant cells is associated with brain tumour progression. Nature 355:846-847, 1992
157. Simon M, von Deimling A, Larson JL, et al: Allelic losses on chromosomes 14, 10, and 1 in atypical and malignant meningiomas: A genetic model of meningioma progression. Cancer Res 55:4696-4701, 1995
158. Slavc I, MacCollin MM, Dunn M, et al: Exon scanning for mutations of the NF2 gene in pediatric ependymomas, rhabdoid tumors and meningiomas. Int J Cancer 64:243-247, 1995
159. Sonoda Y, Iizuka M, Yasuda J, et al: Loss of heterozygosity at 11p15 in malignant glioma. Cancer Res 55:2166-2168, 1995
160. Sonoda Y, Murakami Y, Tominaga T, et al: Delection mapping of chromosome 10 in human glioma. Jpn J Cancer Res 87:363-367, 1996
161. Steck PA, Ligon AH, Cheong P, et al: Two tumor suppressive loci on chromosome 10 involved in human glioblastomas. Genes Chromosomes Cancer 12:255-261, 1995
162. Steck PA, Pershouse MA, Jasser SA, et al: Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nature Genet 15:356-362, 1997
163. Tang P, Steck PA, Yung WKA: The autocrine loop of TGF-a/EGFR and brain tumors. J Neurooncol 35:303-314, 1997
164. Tice H, Barnes PD, Goumnerova L, et al: Pediatric and adolescent oligodendrogliomas. Am J Neuroradiol 14:1293-1300, 1993
165. Tominaga T, Kayama T, Kumabe T, et al: Anaplastic ependymomas: Clinical features and tumour suppressor gene p53 analysis.Acta Neurochir 135:163-170, 1995
166. Trofatter JA, MacCollin MM, Rutter JL, et al: A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor gene. Cell 72:791-800, 1993
167. Tse JYM, Ng H-K, Lau K-M, et al: Loss of heterozygosity of chromosome 14q in low- and high-grade meningiomas. Human Pathol 28:779-785, 1997
168. Tsutsumi K, Kitagawa N, Niwa M, et al: Effect of suramin on 125I-insulin-like growth factor-I binding to human meningiomas and on proliferation of meningioma cells. J Neurosurg 80:502-509, 1994
169. Ueba T, Nosaka T, Takahashi JA, et al: Transcriptional regulation of basic fibroblast growth factor gene by p53 in human glioblastoma and hepatocellular carcinoma cells. Proc Natl Acad Sci USA 91:9009-9013, 1994
170. Ueki K, Ono Y, Henson JW, et al: CDKN2/p16 or RB alterations occur in the majority of glioblastomas and are inversely correlated. Cancer Res 56:150-153, 1996
171. Ueki K, Ramaswamy S, Billings SJ, et al: Chromosomal localization to 19q13.3, partial genomic structure and 5´ cDNA sequence of the human symplekin gene. Somat Cell Mol Genet 23(3): 229-231, 1997
172. Ueki K, Rubio M-P, Ramesh V, et al: MTS1/CDKN2 gene mutations are rare in primary human astrocytomas with allelic loss of chromosome 9p. Hum Mol Genet 3:1841-1845, 1994
173. Van Meyel DJ, Ramsay DA, Casson AG, et al: p53 mutation, expression, and DNA ploidy in evolving gliomas: Evidence for two pathways of progression. J Natl Cancer Inst 86(13):1011-1017, 1994
174. Venter DJ, Thomas DG: Multiple sequential molecular abnormalities in the evolution of human gliomas. Br J Cancer 63:753-757, 1991
175. von Deimling A, Eibl RH, Ohgaki H, et al: p53 mutations are associated with 17p allelic loss in grade II and grade III astrocytoma.Cancer Res 52:2987-2990, 1992
176. von Deimling A, Louis DN, Menon AG, et al: Deletions on the long arm of chromosome 17 in pilocytic astrocytoma. Acta Neuropathol 86:81-85, 1993
177. von Deimling A, Louis DN, Schramm J, et al: Astrocytic gliomas: Characterization on a molecular genetic basis. Recent Results Cancer Res 135:33-42, 1994
178. von Deimling A, Louis DN, von Ammon K, et al: Evidence for a tumor suppressor gene on chromosome 19q associated with human astrocytomas, oligodendrogliomas, and mixed gliomas. Cancer Res 52:4277-4279, 1992
179. von Deimling A, Louis DN, Wiestler OD: Molecular pathways in the formation of gliomas. Glia 15:328-338, 1995
180. von Deimling A, Negel J, Bender B, et al: Deletion mapping of chromosome 19 in human gliomas. Int J Cancer 57:676-680, 1994
181. von Deimling A, von Ammon K, Schoenfeld D, et al: Subsets of glioblastomas multiforme defined by molecular genetic analysis.Brain Pathol 3:19-26, 1993
182. von Haken MS, White ECX, Daneshvar-Shyesther L, et al: Molecular genetic analysis of chromosome arm 17p and chromosome arm 22q DNA sequences in sporadic pediatric ependymomas. Genes Chromosomes Cancer 17:37-44, 1996
183. Wang JL, Zhang ZJ, Hartman M, et al: Detection of TP53 gene mutation in human meningiomas: A study using immunohistochemistry, polymerase chain reaction/single-strand conformation polymorphism and DNA sequencing techniques on paraffin-embedded samples. Int J Cancer 64:223-228, 1997
184. Wang SI, Puc J, Li J, et al: Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res 57:4183-4186, 1997
185. Watanabe K, Tachibana O, Sato K, et al: Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol 6:217-224, 1996
186. Watkins D, Ruttledge MH, Sarrazin J, et al: Loss of heterozygosity on chromosome 22 in human gliomas does not inactivate the neurofibromatosis type 2 gene. Cancer Genet Cytogenet 92:73-78, 1996
187. Weber RG, Sabel M, Reifenberger J, et al: Characterization of genomic alterations associated with glioma progression by comparative genomic hybridization. Oncogene 13:983-994, 1996
188. Weiner HL: The role of growth factor receptors in central nervous system development and neoplasia. Neurosurgery 37:179-194, 1995
189. Wellenreuther R, Kraus JA, Lenartz D, et al: Analysis of the neurofibromatosis 2 gene reveals molecular variants of meningioma.Am J Pathol 146:827-832, 1995
190. Wesseling P, Ruiter DJ, Burger PC: Angiogenesis in brain tumors; pathobiological and clinical aspects. J Neurooncol 32:253-265, 1997
191. Westermark B, Carl-Hendrik H, Nistér M: Platelet-derived growth factor in human glioma. Glia 15:257-263, 1995
192. Wienecke R, Guha A, Maize, Jr., et al: Reduced TSC1 RNA and protein in sporadic astrocytomas and ependymomas. Ann Neurol 42:230-235, 1997
193. Wikstrand CJ, Stanley SD, Humphrey PA, et al: Investigation of a synthetic peptide as immunogen for a variant epidermal growth factor receptor associated with gliomas. J Neuroimmunol 46:165-173, 1993
194. Willert JR, Daneshvar L, Sheffield VC, et al: Deletion of chromosome arm 17p DNA sequences in pediatric high-grade and juvenile pilocytic astrocytomas. Genes Chromosomes Cancer 12:165-172, 1995
195. Wong AJ, Bigner SH, Bigner DD: Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invaribly associated with gene amplification. Proc Natl Acad Sci USA 84:68-99, 1987
196. Wu JK, Folkerth RD, Ye Z, et al: Aggressive oligodendroglioma predicted by chromosome 10 restriction fragment length polymorphism analysis. Case study. J Neurooncol 15:29-35, 1992
197. Yong WH, Chou D, Ueki K, et al: Chromosome 19q deletions in human gliomas overlap telo-meric to D19S219 and may target a 425 kb region centromeric to D19S112. J Neuropathol Exp Neurol 54:622-626, 1995
198. Yong WH, Ueki K, Chou D, et al: Cloning of a highly conserved human protein serine-threonine phosphatase gene from the glioma candidate region on chromosome 19q13.3. Genomics 29:533-536, 1995
199. Zang KD: Cytological and cytogenetical studies on human meningioma. Cancer Genet Cytogenet 6:249-274, 1982
200. Zankl H, Zang KD: Cytological and cytogenetical studies on brain tumors. 4. Identification of the missing G chromosome in human meningiomas as no. 22 by fluorescence technique. Humangenetik 14:167-169, 1972