
Etiology and Epileptogenesis of Hypothalamic Hamartomas: Opening the Door
Stephen W. Coons, MD+
Dawn C. Duane, MD**
Eric W. Johnson, PhD\++
Ronald J. Lukas, PhD*
Jie Wu, MD, PhD§
John F. Kerrigan, MD**#
Divisions of +Neuropathology, **Pediatric Neurology, \Neurology Research, *Neurobiology, and #Comprehensive Epilepsy Center, Barrow Neurological Institute and Children’s Health Center, St. Joseph’s Hospital and Medical Center, Phoenix, Arizona
++Current Address: Molecular Diagnostics and BioBanking, Prevention Genetics, LLC, Marshfield, WI
Abstract
Hypothalamic hamartomas represent a rare but important model of subcortical epileptogenesis. Recent clinical studies, based primarily on intracranial seizure recordings, have established that the hypothalamic hamartoma itself is epileptogenic. At least in some patients, however, the hamartoma contributes to secondary epileptogenesis affecting the neocortex. Seizures arising from either location improve after resection of a hypothalamic hamartoma. This article defines some of the fundamental questions regarding the neurobiology and pathogenesis of this condition, reviews prior scientific studies bearing on basic mechanisms, and proposes new avenues of inquiry that it is hoped will increase our understanding of epilepsy and hypothalamic hamartomas.
Key Words: epilepsy, hypothalamic hamartoma, secondary epileptogenesis
Hypothalamic hamartomas, rare developmental anomalies of the brain, are solitary, congenital, and nonprogressive mass lesions of the hypothalamus. The size of these lesions and their exact location with respect to the hypothalamus vary considerably. An anatomic (surgical) classification scheme based on the local pathological anatomy has been proposed by Valdueza and colleagues. As discussed later in this article, this classification distinguishes between sessile hypothalamic hamartomas (intrinsic to the hypothalamus and often extending into the third ventricle) and pedunculated forms (attached to the inferior aspects of the hypothalamus or tuber cinereum, with a pedicle of variable size.[39]
Clinical symptoms often appear in infancy. Classically, the symptoms associated with hamartomas consist of the triad of seizure disorder, developmental retardation, precocious puberty, or all three. The early seizures are peculiar, with a gelastic (laughing) phenotype, but they often progress to more obvious and more disabling seizures as the child ages. Equally concerning, the child’s cognitive and psychiatric state often (but not always) declines, leading to severe overall functional disability and family and social disruption. Seizures associated with hypothalamic hamartomas are often resistant to medical management.[5]
This article examines some of the fundamental unanswered questions regarding hypothalamic hamartomas, epilepsy, and their association with clinical deterioration in some patients as they age: What are the etiology and pathogenesis of hypothalamic hamartomas? Why is hypothalamic hamartoma tissue intrinsically epileptogenic? What is the nature of the deleterious and sometimes progressive effect of hypothalamic hamartomas (or the resulting seizures) on more remote and widespread areas of brain function? Why do these effects improve in some patients after successful resection of hypothalamic hamartomas?
Epileptogenesis and Hypothalamic Hamartomas
For years, the exact role of hypothalamic hamartomas in seizure onset was undetermined. Several possibilities were considered. First, hypothalamic hamartomas could have merely been markers for abnormalities (and regions of seizure onset) elsewhere in the brain and therefore played no direct role in seizure onset. Second, hypothalamic hamartomas could have caused disordered network relationships that led to seizure onset elsewhere in the brain. Finally, hamartomas could have been the actual site of seizure onset. Earlier concepts that stressed the cortical localization of seizure onset tended to discount the possibility that hypothalamic hamartomas themselves were epileptogenic.[7,10]
During the past 10 years, hypothalamic hamartomas have been shown to be intrinsically epileptogenic. They therefore represent an important and unique human model for subcortical epileptogenesis.[6,11] Contributions to this discovery included studies involving hyperperfusion of hypothalamic hamartomas with ictal single photon emission computed tomography[3,26] and, more importantly, recordings of ictal onset from hypothalamic hamartoma tissue with implanted intracranial recording electrodes.[26,28,30] The final test of this hypothesis, that is, cure of the seizure disorder after resection of hypothalamic hamartomas, has been harder to obtain. Attempts at surgical resection have met with mixed, although sometimes favorable, results. As reviewed by Valdueza and colleagues,[39] many early reports consisted of single cases from a number of centers. Seizures also have responded to other ablative therapies used to treat hypothalamic hamartomas, including Gamma knife radiation[2,38] and stereotactic thermoablation.[31] Furthermore, resective surgery in areas of the brain other than the site of hypothalamic hamartomas has been unsuccessful, even if guided by intracranial ictal electroencephalographic (EEG) monitoring (but not including the hypothalamic hamartoma itself).[13]
Palmini and coworkers[30] found that of 13 patients who underwent resection of a hypothalamic hamartoma,the control of seizures improved in 11. Two of these patients were free of seizures. However, significant complications were associated with the subtemporal surgical approach, including ischemic stroke in four cases. Recently, Rosenfeld and colleagues[33] reported their experience with complete or almost complete anatomic resection of hypothalamic hamartomas through a transcallosal approach. Seizure outcomes were excellent and the rate of complications was low. Further details of the transcallosal approach for hypothalamic hamartoma resection, including preliminary outcomes with this procedure at our institution,[29] are provided elsewhere in this issue.
Although this body of work has established the epileptogenic nature of hypothalamic hamartoma tissue, the story appears to be more complicated. Early in their course, patients with a hypothalamic hamartoma often exhibit gelastic seizures. As these children age, more severe and disabling generalized seizures may develop, often paralleled by a deterioration in cognitive and neuropsychiatric functioning.[5,17] Freeman and colleagues have established the potential for a hypothalamic hamartoma to contribute to secondary epileptogenesis.[19] In the absence of epileptiform activity associated with hamartomas, they found generalized cortical spikewave patterns on intracranial recordings obtained during transcallosal surgery to resect the lesions. After surgery generalized spike-wave patterns on scalp EEGs and clinical tonic seizures persisted. As long as 6 months after surgery, the tonic seizures improved and eventually disappeared in 11 of 12 patients.[19] This “running down” phenomenon has been observed previously in patients with neocortical (temporal lobe) epilepsy and appears to correlate with the percent resection of the epileptogenic zone.[34] The mechanisms that may account for this running-down phenomenon in subcortical epilepsies remain unknown.
Neuropathology and Neurobiology of Hypothalamic Hamartomas
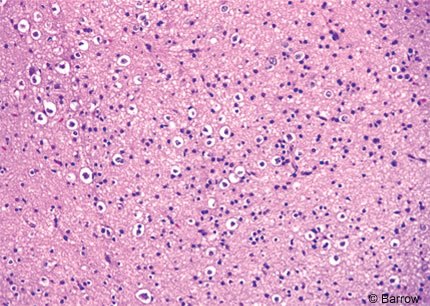
Articles about the imaging features of hypothalamic hamartomas have been published.[1] However, little information is available about the basic neuropathological or neurobiological underpinnings of hypothalamic hamartomas and epilepsy.[20] Some standard neuropathology texts do not even mention hypothalamic hamartomas.
By definition, a hamartoma consists of a disorderly overgrowth of mature and normal-appearing cells. A standard neuropathological examination of resected hypothalamic hamartoma tissue shows relatively normal-appearing neuronal elements (Fig. 1). The surrounding neuropil often appears somewhat spongy. Hypothalamic hamartomas lack the enlarged or “balloon” cells characteristic of focal cortical dysplasias and other pathologies such as tuberous sclerosis. The degree of cellularity varies from case to case, but anaplastic elements are not seen. Neurons can be grouped into variably demarcated nodules, but these lesions lack the laminar architecture and relationship with white matter tracts that characterize normal brain. Unorganized glial elements, including astrocytes and oligodendrocytes, are diffusely distributed in the lesions but are seldom prominent.
Hypothalamic hamartomas associated with precocious puberty may stain for gonadotrophin-releasing hormone (GnRH).[15] One hypothesis is that hypothalamic hamartomas directly secrete GnRH, acting as an ectopic GnRH pulse generator, independent of the usual central inhibitory mechanisms. 27 An alternative hypothesis is that hypothalamic hamartomas do not release GnRH but rather elaborate transforming growth factor-a (TGF-(alpha)) from astroglial cells. In turn, the TGF-(alpha) acts as a positive trophic factor for GnRH release from normal hypothalamic tissue.[22,24] Regardless of the underlying mechanism, patients who manifest with precocious puberty appear to respond to GnRH agonists, which presumably operate by negative feedback.[16] Curiously, the gelastic seizures of one patient were reported to cease after GnRH-agonist therapy.[42]
Although the classic symptom triad of hypothalamic hamartomas consists of gelastic seizures, developmental delay, and precocious puberty, it is increasingly recognized that overlap between children presenting with precocious puberty and those presenting with epilepsy is incomplete.[1,23] In most patients with precocious puberty, their hypothalamic hamartoma occupies a “parahypothalamic” position with inferior or even pedunculated anatomy relative to the hypothalamus. Few of these patients develop epilepsy (Type Ia and Ib as classified by Valdueza et al.).[39] Conversely, in most patients with epilepsy, the hamartoma occupies an “intrahypothalamic” anatomic position, often extending into the third ventricle. Most of these patients do not develop precocious puberty (Type IIa and IIb as classified by Valdueza et al.).[39]
Studies relating to the neuropathology and neurobiology of hypothalamic hamartomas associated with epilepsy are extremely limited. A study of 13 resected cerebral hamartomas (none arising from the hypothalamus) associated with refractory epilepsy documented that the lesions are pathologically nonproliferative based on mitotic counts and negative immunohistochemistry for a nuclear marker of cell division (MIB-1 antibody).[40] This result is not surprising because serial imaging studies show hamartomas to be stable and nonprogressive. Valdueza and colleagues[39] performed immunohistochemical studies with an extensive panel of neuronal and neurosecretory antibodies in a small series of patients with a hypothalamic hamartoma who had epilepsy. All resected tissue specimens were positive for neuronal markers, including neuron-specific enolase, synaptophysin, and neurofilament protein. All specimens were negative for luteinizing hormone-releasing hormone (LHRH, also known as GnRH). None of these patients had precocious puberty. Two of three specimens tested were positive for other neuropeptides (despite the lack of evidence for an endocrine disturbance), including corticotrophin-releasing factor (CRF) and met-enkephalin. The significance of these findings is unknown. However, CRF has been proposed as a proconvulsant neuropeptide in a leading hypothesis relating to the pathogenesis of infantile spasms.[4]
Most hypothalamic hamartomas are sporadic and not associated with a positive family history or identifiable syndromes. However, about 10% of hypothalamic hamartomas are associated with Pallister-Hall syndrome, an autosomal dominant malformation syndrome that includes postaxial polydactyly, imperforate anus, and a bifid epiglottis, among other congenital anomalies.[21,35] The Pallister-Hall syndrome has been associated with a germ-line defect in the Gli3 gene,[25] a zinc-finger transcription factor protein in the sonic hedgehog pathway.[36] The possibility that a somatic mutation of Gli3 may be responsible for sporadic cases of hypothalamic hamartomas is under investigation (Robyn Wallace, personal communication, 2003).
The original report of the neuropathology associated with the Pallister-Hall syndrome described histological features designated as hamartoblastoma.[14] Subsequent reports, however, have found no significant differences in the neuropathology associated with Pallister-Hall cases and those of patients with sporadic hypothalamic hamartomas. [6,35] An animal model with a Gli3 knockout mutation has been reported but is not associated with hypothalamic hamartomas.[9]
Current Research Prospects
Barrow has established a multidisciplinary center for the evaluation and management of hypothalamic hamartomas. Available treatments include transcallosal surgical resection of hypothalamic hamartomas. Access to such tissue from multiple patients at a single institution enables concentrated research efforts. It is providing an opportunity to study basic issues related to the pathogenesis of these lesions and their intrinsic epileptogenesis. It is hoped that these investigations will offer additional insights into the mechanisms of secondary epileptogenesis. This review identifies current areas of research interest. Original observations resulting from these research efforts will be submitted for publication in peer-reviewed journals.
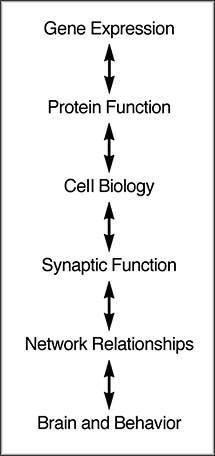
Specific research questions can be organized based on their increasing biological complexity (Fig. 2). Each level of this hierarchy presents questions and challenges for a problem such as hypothalamic hamartomas with its associated symptoms. We hope to exploit multiple lines of inquiry to make progress in answering fundamental questions about hypothalamic hamartomas.
Cell Cultures
Primary explant or dissociated cell cultures present a snapshot of the cellular profile occurring in a lesion in vivo. Cells emanating from a tissue explant exhibit a migratory capacity that may be informative of their phenotype. An advantage of dissociated cell cultures is the ready visibility of cell morphology. Even postmitotic cells, such as neurons, can be coaxed to survive for a few days in primary cell culture media rich with nutrients and growth factors. Samples can be stained using immunocytochemical techniques to assess cellular phenotypes (i.e., neuronal, glial, other) based on their expression of cell type-specific markers. Skilled investigators also can distinguish cell types based on morphology.
To the extent that some cells in the primary culture are capable of division, their numbers can be multiplied and their phenotypes can be established based on their morphological and staining characteristics. Any cells that continue to proliferate may reach the criteria for a continuous cell-line culture. Such longer-term cultures allow the presence of immortalized, tumorlike cells or cells with proliferative capacity to be assessed.
Explant and dissociated cell cultures can be used to test different hypotheses about the content and origin of hypothalamic hamartomas. Given that hypothalamic hamartomas are nonprogressive mass lesions (sometimes quite large), they defy easy categorization as dysplasias or neoplasms. However, current concepts of pathogenesis for these two broad categories of disease can be used to generate hypotheses about the origin of hypothalamic hamartomas.
One hypothesis is that hypothalamic hamartomas may result from ectopic localization of otherwise relatively normal cellular elements. Physical misplacement of hypothalamic hamartomas tissue could be related to defects in cell-cell recognition, which normally guides the sequential processes of neuroepithelial proliferation, differentiation of primitive neuroectodermal cells to neurons and glia, and postmitotic neuronal migration along radial glial cells.
Misplacement in space also implies the possibility of misplacement in time. Cell-to-cell signaling events that accompany a sequence of developmentally determined windows of brain development may be aberrant, thereby contributing to the organization of abnormal brain tissue as seen with hypothalamic hamartomas. These signaling mechanisms may result from soluble ligand-receptor interactions or from abnormalities of substrate-cell interaction. For example, the premature or late arrival of migratory signals when the surrounding tissue is no longer permissive for movement of cells in response to those signals would lead to abnormal local brain structure.
A second hypothesis (also greatly simplified) is that hypothalamic hamartoma cells (or, perhaps more likely, one cellular constituent of hypothalamic hamartoma tissue) are positioned normally but have an abnormal proliferative potential. Immunohistochemical studies focusing on cell lineage may identify that the hypothalamic hamartoma cells themselves are intrinsically abnormal, potentially expressing more primitive cell markers or markers representative of more than one cell line. Studies of cell cultures may be capable of identifying such abnormalities, including “tumor-like” and “stem-cell like” phenotypes.
These lines of investigation are not likely to be mutually exclusive because the neurobiology of cellular proliferation, cellular destination, and tissue integration reflects complex and mutually dependent interactions. A starting hypothesis may be that all cases of sporadic hypothalamic hamartoma share the same mechanism. Although by no means assured, a careful search for variability within the tissue phenotype of sporadic hypothalamic hamartomas is important to detect clues that may lead in different directions. Furthermore, empirically based discoveries leading to the formulation of hypotheses may be the most appropriate route for studies of comparatively unknown entities such as hypothalamic hamartomas.
Are hypothalamic hamartomas reflective of a somatic mutation in an early neuroepithelial precursor? Such a mutation might account for either a migrational or proliferative mechanism. Cells in some, if not all, brain regions originate clonally from a single primitive neuroectodermal precursor, maturing into either glial or neuronal cells. Even if there is no genomic (germ-line) mutation that would be carried by all cells in the body, the appearance of a somatic mutation confined to such a precursor could give rise to a cadre of glial and neuronal cells that would carry that mutation into their progeny, distinct from cells elsewhere in the brain.
Gene Expression and Genetic Studies
Whether hypothalamic hamartomas are caused by developmental abnormalities that alter cellular profiles or physical position within the lesion, differences in gene expression as messenger ribonucleic acid (mRNA) and protein may be responsible. For example, abnormalities of gene expression causing either quantitative or qualitative abnormalities of cellular proteins involved in cell-cell recognition, migration, or chemical or electrical signaling could lead to abnormal organization of the brain at the lesion site.
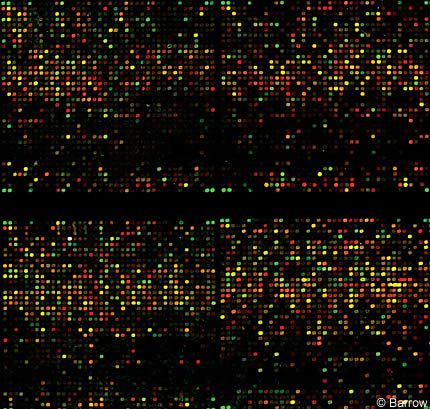
One technique of identifying unique patterns of gene expression characteristic of hypothalamic hamartomas involves the use of microarrays of deoxyribonucleic acid (DNA, gene chips).[12,18,37] In this approach, mRNA is extracted and converted to copy or complementary DNA (cDNA) through a process designed to preserve initial ratios of expression of differentmRNAs. Gene expression in hypothalamic hamartoma tissue and a control specimen is then compiled. In one sensitive and commonly applied technique, cDNAs prepared from each sample are labeled with different fluorescent dyes. The mixed samples are allowed to compete for binding sites on anti-sense cDNAs or oligonucleotide immobilized on the gene chip. If a particular gene in hypothalamic hamartoma tissue is overexpressed compared to a control specimen, more of the labeled cDNA from that specimen will bind to the gene chip. Conversely, under expression of the gene in the diseased specimen will be reflected by higher labeling with control cDNA (Fig. 3).
Given knowledge of the map of immobilized cDNA or oligonucleotides on the microarray chip, the identity of the gene with altered expression can be determined. Binding sites for nonoverlapping segments of the same gene on the microarray chip can verify the result. Candidates identified based on this gene expression mapping are subsequently subjected to independent verification and validation using other technologies, such as real-time, quantitative reverse transcription-polymerase chain reactions (PCR), mRNA in-situ hybridization, or immunocytochemistry.
These approaches could be applied to bulk tissue samples of hypothalamic hamartomas for comparison to the best possible control brain tissues. Because of major challenges in defining the best control specimen for samples in bulk, the same techniques could be applied to explant or dissociated cultures, potentially enriched for the same cell type, or even to cellular subsets harvested by laser capture.
Differences in gene expression as mRNA between hypothalamic hamartomas and normal tissues could be related to somatic or genomic mutations. If so, gene expression profiling through DNA microarray analyses has the potential to identify the responsible genes. In some instances, gene expression profiling may appear normal, but mRNAs and encoded proteins may nevertheless be abnormal due to gene mutations. Invariably, gene sequences are polymorphic, meaning that they differ between individuals, often subtly. In many cases, a polymorphism is not mutagenic: The protein encoded by the gene in question is not abnormal, but the polymorphism is a marker for some other genetic defect. Polymorphisms can occur in DNA sequences not involved in coding for proteins. Nevertheless, they could affect expression of nearby genes, serve as markers for gene mutations or polymorphisms, or both. Because of these possibilities, more global genetic screening of hypothalamic hamartoma samples is warranted in addition to gene expression profiling.
Genomic mutations carried by every cell in the body can be revealed by a number of different genetic screening approaches: direct sequencing of genomic DNA from readily available peripheral tissues (lymphocytes or skin) or comparing affected to unaffected individuals to determine if specific mutations or polymorphisms occur with a higher frequency in individuals with hypothalamic hamartomas. Among other options for higher-throughput screening for polymorphisms are restriction fragment length polymorphism (RFLP) analysis, a test commonly used in crime laboratories, and single nucleotide polymorphism (SNP) mapping. The latter is based on new and emerging knowledge about common polymorphisms in the human genome and is designed to focus attention on the fewer sites of sequence difference rather than the many more uninformative areas of sequence identity across individuals.
Similar experiments could be applied to verify somatic mutations occurring in only a subset of cells in the body. Such approaches would need to be well designed to take advantage of banked hypothalamic hamartoma specimens and to be informed by the results of studies of related specimens. For example, if a somatic mutation was present in a subset of neuronal cells in hypothalamic hamartomas but not control tissue and not in non-neuronal hypothalamic hamartomas cells, then SNP mapping would be most optimal if confined to cDNA derived from neuronal cells. If functional studies identify a potentially defective gene, single gene reverse transcription-PCR analysis followed by gene sequencing could be used to identify the mutation.
In cases of familial hypothalamic hamartomas, any form of mapping of the evident genomic mutation would be enhanced by within-family comparisons. Such studies could rapidly lead to the identification of genes and specific mutations.
Cellular Physiology of Hypothalamic Hamartoma Neurons
The common manifestation of hypothalamic hamartomas with epileptic seizures implies a defect in neuronal chemical and/or electrical signaling, perhaps influenced or accompanied by an abnormality in glial regulation of that signaling. The availability of primary explants and acutely dissociated cells from hypothalamic hamartoma tissue affords the opportunity to ascertain whether the electrophysiological characteristics of cells from the lesion site differ from those of comparable cells from normal control brain. Clues about possible molecular mechanisms for the intrinsic epileptogenesis of hypothalamic hamartoma tissue derive from existing knowledge about molecular mechanisms involved in other forms of epilepsy.
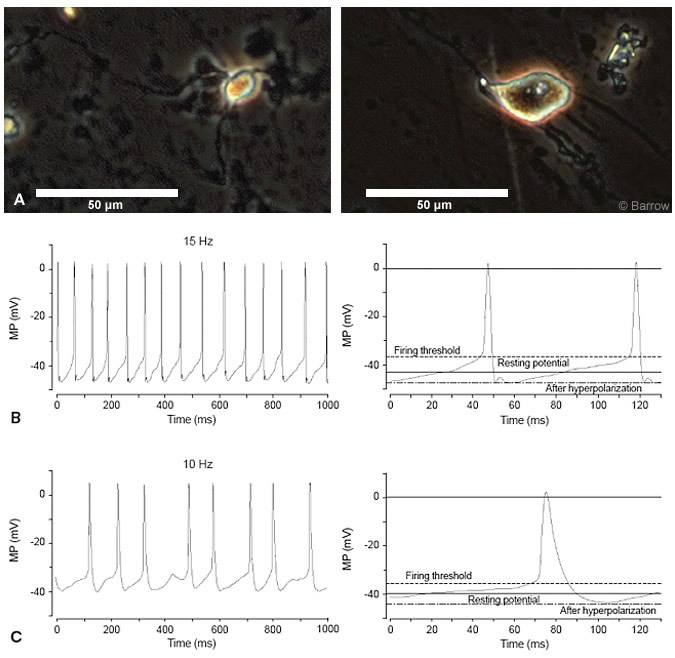
One approach is to generate acutely dissociated cells from hypothalamic hamartoma tissue. The powerful approach of patch clamp electrophysiology could then be used to characterize the electrical properties and ligand sensitivity of these cells. The procedure for acutely dissociating cells from human hypothalamic hamartoma tissue is a modification of an existing technique.[41] Separated cells usually adhere to the bottom of the culture dish within 30 minutes and are classified as small (90% of the total cells in hypothalamic hamartoma samples) or large (Fig. 4A).
Logically, attention should first be directed to determining whether cells derived from hypothalamic hamartomas have electrical properties characteristic of glia or neurons. Evidence of spontaneous electroexcitability or of voltage jump-induced transmembrane conductance changes would be indicative of the expression of voltagegated ion channels. Because the activation and inactivation kinetics and details of voltage sensitivity for many voltage-gated sodium, potassium, and calcium channels have been characterized, specific features of such findings in hypothalamic hamartomas would be informative about cellular ionchannel phenotype.
Abnormalities that may be present in hypothalamic hamartoma neurons and that would help explain epileptogenesis would include delayed inactivation kinetics for voltage-gated sodium channels or altered voltage-gated potassium-channel kinetics. These possibilities are raised by the evolving view that many of the epilepsies identified by molecular mechanisms are “channelopathies,” reflecting the abnormal function of ion channels.
Both small and large acutely dissociated hypothalamic hamartomas cells are characterized by a pacemaker-like depolarization that spontaneously brings the membrane potential from the trough of the afterspike hyperpolarization to its firing threshold (Fig. 4B and C). This pattern is similar to previously described pacemaker single-spike activity in rat hypothalamic neurons.[8,32] More detailed studies of these activities are in progress, but the expression of pacemaker-like singlespike activity may contribute to epileptogenesis in patients with hypothalamic hamartomas.
Ligand-gated ion channel function also could be abnormal in hypothalamic hamartomas. Altered properties of gamma amino butyric acid (GABA) or glutamate-activated ion channels are possible given the prime roles of glutamate and GABA in excitatory and inhibitory neurotransmission, respectively. The role of nicotinic acetylcholine receptor subunit mutations in some hereditary epilepsies, also characterized by the onset of seizures in children, has been established. Information is accumulating on the properties of these and other ligand-gated ion channels, and the search for abnormalities involving these receptor-channel complexes in hypothalamic hamartoma tissue may be rewarding.
If patch clamp studies suggest defects in voltage- or ligand-gated ion channels, an obvious line of study would be to determine if such defects originate in genomic (especially in cases within families) or somatic gene mutations.
Conclusion
The ability to pursue basic investigations into the pathogenesis and intrinsic epileptogenesis of hypothalamic hamartomas has been limited by the rarity of these lesions and the relatively scarce amounts of tissue at a single research center. The recently developed transcallosal approach for the surgical resection of hypothalamic hamartomas represents a major step forward in the clinical management of patients affected by this disease. Concomitantly, the availability of tissue from this procedure offers an unprecedented opportunity for understanding this unique form of subcortical epilepsy and secondary epileptogenesis.
References
- Arita K, Ikawa F, Kurisu K, et al: The relationship between magnetic resonance imaging findings and clinical manifestations of hypothalamic hamartoma. J Neurosurg 91:212-220, 1999
- Arita K, Kurisu K, Iida K, et al: Subsidence of seizure induced by stereotactic radiation in a patient with hypothalamic hamartoma. Case report. J Neurosurg 89:645-648, 1998
- Arroyo S, Santamaria J, Sanmarti F, et al: Ictal laughter associated with paroxysmal hypothalamopituitary dysfunction. Epilepsia 38:114-117, 1997
- Baram TZ: Pathophysiology of massive infantile spasms: Perspective on the putative role of the brain adrenal axis. Ann Neurol 33:231-236, 1993
- Berkovic SF, Andermann F, Melanson D, et al: Hypothalamic hamartomas and ictal laughter: Evolution of a characteristic epileptic syndrome and diagnostic value of magnetic resonance imaging. Ann Neurol 23:429-439, 1988
- Berkovic SF, Arzimanoglou A, Kuzniecky R, et al: Hypothalamic hamartoma and seizures: a treatable epileptic encephalopathy. Epilepsia 44:969-973, 2003
- Berkovic SF, Kuzniecky RI, Andermann F: Human epileptogenesis and hypothalamic hamartomas: New lessons from an experiment of nature. Epilepsia 38:1-3, 1997
- Beurrier C, Bioulac B, Hammond C: Slowly inactivating sodium current (I(NaP)) underlies single-spike activity in rat subthalamic neurons. J Neurophysiol 83:1951-1957, 2000
- Bose J, Grotewold L, Ruther U: Pallister-Hall syndrome phenotype in mice mutant for Gli3. Hum Mol Genet 11:1129-1135, 2002
- Breningstall GN: Gelastic seizures, precocious puberty, and hypothalamic hamartoma. Neurology 35:1180-1183, 1985
- Breningstall GN: Gelastic seizures, precocious puberty, and hypothalamic hamartoma. Reply to letter. Neurology 36:444, 1986
- Butte A: The use and analysis of microarray data. Nat Rev Drug Discov 1:951-960, 2002
- Cascino GD, Andermann F, Berkovic SF, et al: Gelastic seizures and hypothalamic hamartomas: evaluation of patients undergoing chronic intracranial EEG monitoring and outcome of surgical treatment. Neurology 43:747-750, 1993
- Clarren SK, Alvord EC, Jr., Hall JG: Congenital hypothalamic hamartoblastoma, hypopituitarism, imperforate anus, and postaxial polydactyly–a new syndrome? Part II: Neuropathological considerations. Am J Med Genet 7:75-83, 1980
- Culler FL, James HE, Simon ML, et al: Identification of gonadotropin-releasing hormone in neurons of a hypothalamic hamartoma in a boy with precocious puberty. Neurosurgery 17:408-412, 1985
- de Brito VN, Latronico AC, Arnhold IJ, et al: Treatment of gonadotropin dependent precocious puberty due to hypothalamic hamartoma with gonadotropin releasing hormone agonist depot. Arch Dis Child 80:231-234, 1999
- Deonna T, Ziegler AL: Hypothalamic hamartoma, precocious puberty and gelastic seizures: a special model of “epileptic” developmental disorder. Epileptic Disord 2:33-37, 2000
- Dobrin SE, Stephan DA: Integrating microarrays into disease-gene identification strategies. Expert Rev Mol Diagn 3:375-385, 2003
- Freeman JL, Harvey AS, Rosenfeld JV, et al: Generalized epilepsy in hypothalamic hamartoma: Evolution and postoperative resolution. Neurology 60:762-767, 2003
- Graham DI, Lantos PL: Regional neuropathology: Hypothalamus and pituitary, in Graham DI, Lantos PL (eds): Greenfield’s Neuropathology. London: Arnold, 1997, pp 1007-1094
- Hall JG, Pallister PD, Clarren SK, et al: Congenital hypothalamic hamartoblastoma, hypopituitarism, imperforate anus and postaxial polydactyly–a new syndrome? Part I: Clinical, causal, and pathogenetic considerations. Am J Med Genet 7:47-74, 1980
- Jung H, Carmel P, Schwartz MS, et al: Some hypothalamic hamartomas contain transforming growth factor alpha, a puberty-inducing growth factor, but not luteinizing hormone-releasing hormone neurons. J Clin Endocrinol Metab 84:4695-4701, 1999
- Jung H, Neumaier PE, Hauffa BP, et al: Association of morphological characteristics with precocious puberty and/or gelastic seizures in hypothalamic hamartoma. J Clin Endocrinol Metab 88:4590-4595, 2003
- Jung H, Ojeda SR: Pathogenesis of precocious puberty in hypothalamic hamartoma. Horm Res 57 Suppl 2:31-34, 2002
- Kang S, Graham JM, Jr., Olney AH, et al: GLI3 frameshift mutations cause autosomal dominant Pallister-Hall syndrome. Nat Genet 15:266-268, 1997
- Kuzniecky R, Guthrie B, Mountz J, et al: Intrinsic epileptogenesis of hypothalamic hamartomas in gelastic epilepsy. Ann Neurol 42:60-67, 1997
- Mahachoklertwattana P, Kaplan SL, Grumbach MM: The luteinizing hormone-releasing hormone-secreting hypothalamic hamartoma is a congenital malformation: Natural history. J Clin Endocrinol Metab 77:118-124, 1993
- Munari C, Kahane P, Francione S, et al: Role of the hypothalamic hamartoma in the genesis of gelastic fits (a video-stereo-EEG study). Electroencephalogr Clin Neurophysiol 95:154-160, 1995
- Ng Y-T, Kerrigan JF, Rekate HL, et al: Transcallosal resection of hypothalamic hamartoma in the treatment of refractory epilepsy (abstract). Epilepsia 44 (Suppl 9):158-159, 2003
- Palmini A, Chandler C, Andermann F, et al: Resection of the lesion in patients with hypothalamic hamartomas and catastrophic epilepsy. Neurology 58:1338-1347, 2002
- Parrent AG: Stereotactic radiofrequency ablation for the treatment of gelastic seizures associated with hypothalamic hamartoma. Case report. J Neurosurg 91:881-884, 1999
- Pennartz CM, Bierlaagh MA, Geurtsen AM: Cellular mechanisms underlying spontaneous firing in rat suprachiasmatic nucleus: Involvement of a slowly inactivating component of sodium current. J Neurophysiol 78:1811-1825, 1997
- Rosenfeld JV, Harvey AS, Wrennall J, et al: Transcallosal resection of hypothalamic hamartomas, with control of seizures, in children with gelastic epilepsy. Neurosurgery 48:108-118, 2001
- Salanova V, Andermann F, Rasmussen T, et al: The running down phenomenon in temporal lobe epilepsy. Brain 119 ( Pt 3):989-996, 1996
- Schaefer GB, Olney AH: Hypothalamic dysfunction with polydactyly and hypoplastic nails. Semin Pediatr Neurol 6:238-242, 1999
- Shin SH, Kogerman P, Lindstrom E, et al: GLI3 mutations in human disorders mimic Drosophila cubitus interruptus protein functions and localization. Proc Natl Acad Sci U S A 96:2880-2884, 1999
- Sturla LM, Fernandez-Teijeiro A, Pomeroy SL: Application of microarrays to neurological disease. Arch Neurol 60:676-682, 2003
- Unger F, Schrottner O, Haselsberger K, et al: Gamma knife radiosurgery for hypothalamic hamartomas in patients with medically intractable epilepsy and precocious puberty. Report of two cases. J Neurosurg 92:726-731, 2000
- Valdueza JM, Cristante L, Dammann O, et al: Hypothalamic hamartomas: With special reference to gelastic epilepsy and surgery. Neurosurgery 34:949-958, 1994
- Volk EE, Prayson RA: Hamartomas in the setting of chronic epilepsy: A clinicopathologic study of 13 cases. Hum Pathol 28:227-232, 1997
- Wu J, Chan P, Schroeder KM, et al: 1-methyl-4-phenylpridinium (MPP+)-induced functional run-down of GABA(A) receptor-mediated currents in acutely dissociated dopaminergic neurons. J Neurochem 83:87-99, 2002
- Zaatreh M, Tennison M, Greenwood RS: Successful treatment of hypothalamic seizures and precocious puberty with GnRH analogue. Neurology 55:1908-1910, 2000