
Symptomatic Seizures and Epilepsy Related to Hypothalamic Hamartomas
Yu-tze Ng, MD
John F. Kerrigan, MD
Divisions of Pediatric Neurology and Comprehensive Epilepsy Center, Barrow Neurological Institute and Children’s Health Center, St. Joseph’s Hospital and Medical Center, Phoenix, Arizona
Abstract
Refractory, mixed seizure disorder and epilepsy related to hypothalamic hamartomas are usually the most devastating symptomology that afflicts patients and their families. Typically, patients experience different seizure types, including characteristic gelastic seizures, which are refractory to most anti-epileptic drugs. Seizures cause significant disability, co-existing with developmental disturbances and neuropsychiatric symptoms, and may contribute to the progressive deterioration in cognition and behavior seen in some patients. Previously it was uncertain if hypothalamic hamartomas were responsible for the seizures and whether resection of the lesions would improve the seizures. Recently, both of these hypotheses have been proven correct.
Key Words: seizures, epilepsy, hypothalamic hamartoma, transcallosal resection
Hamartomas of the hypothalamus and tuber cinereum are congenital malformations consisting of tumorlike masses of neuronal tissue in an ectopic location.[7] Hypothalamic hamartomas are rare although no data have been published on their prevalence or incidence. Anecdotally, their prevalence is estimated to be about one in a million. It may be that the prevalence is underestimated because these lesions can go undetected without high-resolution magnetic resonance (MR) imaging. Various symptoms, predominantly symptomatic seizures and epilepsy, isosexual precocious puberty, intellectual impairment, and behavioral problems, have been associated with hypothalamic hamartomas. [2,7]
Seizure Types and Localization
Although most patients tend to have multiple seizure types (mixed seizure disorder), the most characteristic seizure type is gelastic (laughing) seizures, which were first described by Daly and Mulder in 1957.[6] Gelastic seizures often are the earliest seizure type manifested by patients with hypothalamic hamartomas, often during the first days of life.[9] The brief, ictal phase of the laughter, smile, or both may be disregarded by primary care physicians, and the condition may not be diagnosed for months or even years (sometimes only when other seizure types manifest). Gelastic seizures have a variable phenotype, sometimes occurring with intermixed elements of laughing, crying, or fearfulness. Typically, they are brief, often just a few seconds in duration, but they occur frequently, with multiple daily events.
The sessile or intrahypothalamic type, in which the hypothalamic hamartoma is enveloped by the hypothalamus, is most strongly associated with epilepsy. This association probably reflects their anatomic position in or adjacent to central pathways that may mediate seizure activity, such as connections to limbic circuitry.[1,19] Hamartomas also arise from the inferior surface of the hypothalamus (parahypothalamic type), and their morphology is often pedunculated. Such hypothalamic hamartomas are more strongly associated with precocious puberty. Co-existing epilepsy is rare.
Recently, evidence about the origin of different seizure types in patients with hypothalamic hamartomas has been accumulating. Video-electroencephalography (EEG) depth electrode studies, including electrodes inserted into the hamartoma itself as well as into other candidate regions for seizure onset, have helped localize the seizure types. In addition, hyperperfusion of the hypothalamic hamartoma with ictal events has been observed. Together, these studies demonstrate that the hypothalamic hamartoma itself is intrinsically epileptogenic.[4,9,11-13,19]
Over time, and particularly between the ages of 4 and 10 years, hypothalamic hamartoma patients usually begin to have multiple types of seizures. Although almost all seizure types have been observed, they typically include tonic, tonic-clonic, and complex partial seizures with clinical and EEG features of a secondary generalized epilepsy. [2,9,19]
Freeman et al.[9] have proposed that gelastic seizures arise directly from the hypothalamic hamartoma and that the other seizure types evolve with time as the rest of the brain becomes secondarily epileptogenic (able to generate seizures independently). In selected cases, intracranial electrode recordings have documented spike-wave patterns arising from neocortical (frontal) regions in the absence of simultaneous ictal activity in the hypothalamic hamartoma itself. This finding requires further confirmation and refinement, but it may help to explain the observation that other seizure types develop months or years after the onset of gelastic seizures. The mechanisms of secondary epileptogenesis associated with subcortical epilepsy (or other forms of epilepsy) are unknown.
Theoretically, the neocortex “learns” to generate seizures independently (i.e., becomes secondarily epileptogenic) after constant exposure to the repetitive seizures caused by the hypothalamic hamartoma. Alternatively, over time, the hypothalamic hamartoma tissue (regardless of the presence or absence of seizures arising from the hamartoma) may alter network relationships in such a way as to lead to secondary epileptogenesis in cortical regions.
After surgical resection of a hypothalamic hamartoma, many patients immediately become free of seizures. However, some patients continue to have seizures in the immediate and intermediate postoperative period. Presumably, if these patients’ hypothalamic hamartoma has been resected successfully, the seizures persist from neocortical regions affected by the process of secondary epileptogenesis.
Fortunately, there also appears to be a process of “running down” of the epileptogenic potential of the neocortex. In most patients seizure activity improves or ceases completely within 6 months.[9] This process of “unlearning” implies that the hypothalamic hamartoma has a dynamic or ongoing distant effect on neocortex, rather than inciting some form of static change with permanent residual effects. The cellular and molecular mechanisms by which this process occurs are of tremendous interest. On a practical level, postoperative seizures in the first several months after surgical resection of the hypothalamic hamartoma do not necessarily represent surgical failure. Successful seizure control may simply be delayed in some patients.
Seizure Monitoring
Traditional standards for evaluating patients for potential epilepsy surgery include scalp video-EEG monitoring to localize the region or regions of seizure onset. Seizures are recorded and evaluated in detail in terms of both seizure phenotype and maximal electrical field expression of the ictal rhythm involving the scalp electrodes. Nonlesional cases (patients without an abnormality apparent on high-resolution MR imaging such as mesial temporal sclerosis or focal cortical dysplasia) usually require additional seizure monitoring with intracranial electrodes. Depending on the specifics of the individual case, the additional monitoring may include implantation of intracranial depth recording wires, subdural grids, or both.
We believe that epilepsy patients with hypothalamic hamartomas may represent a qualified exception to the standard practice of obtaining preoperative video-EEG monitoring. Among patients with hypothalamic hamartomas, routine scalp video-EEG monitoring fails to localize the site of seizure onset and can suggest that seizures may be arising from cortical structures, such as the temporal, frontal, or even occipital lobes. Accordingly, scalp video-EEG monitoring can actually be misleading in terms of localizing the onset of seizures. Cascino and colleagues have documented this experience in eight patients with refractory epilepsy and hypothalamic hamartomas.[5] These patients underwent a standard preoperative evaluation, including scalp video-EEG monitoring. Seizure onset was localized to the temporal lobe in seven patients and to the frontal lobe in one. None of the patients improved after resection of temporal or frontal tissue. We have had an identical experience with one of our patients in the pretranscallosal series. Recently, one of our patients underwent a successful transcallosal resection after a previously unsuccessful frontal lobe procedure.
Hypothalamic hamartomas are now understood to be intrinsically epileptogenic. Consequently, the failure of scalp EEG to localize seizure onset accurately to these deep near-midline lesions is unsurprising. It also appears that neo-cortical seizure onset in patients affected by secondary epileptogenesis depends on the presence of the hamartoma. It is resection of the hamartoma, rather than of the cortical regions, that ultimately controls seizures in most of these patients. Accordingly, even in these cases, scalp video-EEG recording is of limited value.
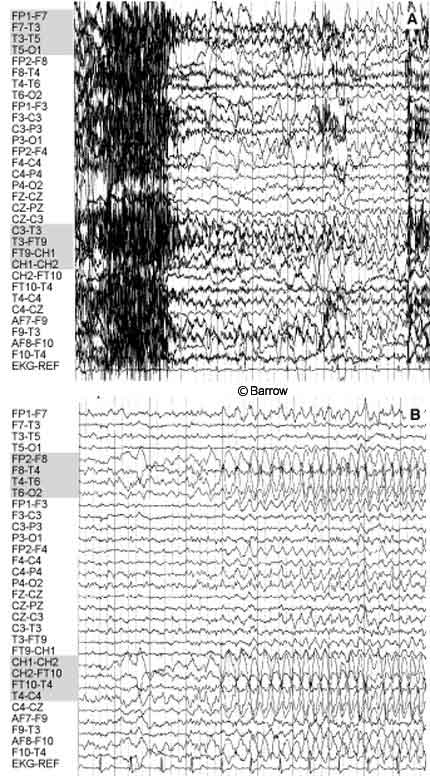
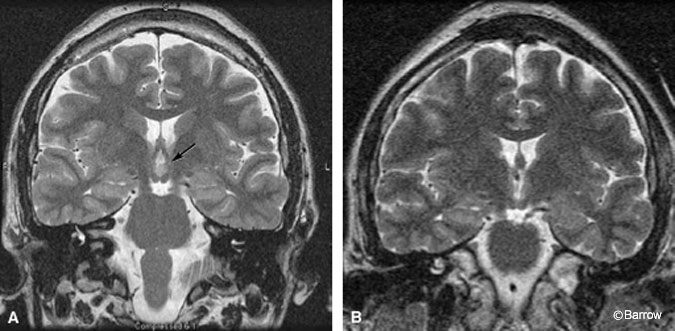
Scalp monitoring may suggest multifocal onset in patients who subsequently undergo successful resection of a hypothalamic hamartoma. For example, the scalp-EEG recordings of the onset of two different complex partial seizures with similar semiology in a 30-year-old man showed that the onset of one seizure appeared to originate from the left temporal region (Fig. 1A) while a second seizure seemed to start from the right temporal region (Fig. 1B). During the patient’s inpatient stay in the epilepsy monitoring unit, several seizures were recorded. Most of the seizures appeared to originate from one temporal lobe or the other (three from the left temporal lobe, one from the right temporal lobe, and two nonlocalizing). The patient also had frequent, bilateral, independent, interictal temporal lobe epileptiform discharges. The patient was determined to have a hypothalamic hamartoma (Fig. 2A). Endoscopic resection of the hamartoma was followed by complete seizure control during the first few weeks of followup (Fig. 2B).
Preoperative seizure monitoring is required when other cerebral pathology co-exists on MR imaging and there are questions about the cause of certain spells reported by the patient or family, or if the frequency of epileptic seizures needs to be confirmed. In terms of localization, we believe that seizure monitoring with scalp electrodes offers no information that ultimately assists surgical decision-making. In some cases, it may even be misleading and counterproductive. When such information is important to surgical decision-making, definitive seizure localization in patients with a hypothalamic hamartoma requires intracranial monitoring, including depth wire placement into the hamartoma.
Treatment of Seizures and Epilepsy
Gelastic and other types of seizures associated with hypothalamic hamartomas are notoriously difficult to treat with traditional or even newer antiepileptic drugs (AEDs). Despite high doses of multiple AEDs, freedom from seizures or even good seizure control is rarely achieved in this group of patients. Other therapies used to treat the seizures–vagal nerve stimulation, Gamma knife therapy, stereotactic destruction of the lesion through radiofrequency, and even gonadotrophinreleasing hormone (GnRH) analogue–have had limited success.[8,10,12,14-18,20]
Previously, surgical resection of a hypothalamic hamartoma was undertaken reluctantly. It was thought that hypothalamic hamartomas were not clearly responsible for the seizures and that these lesions were inaccessible and too risky to try to resect. Increasing evidence now indicates that hypothalamic hamartomas can indeed be treated effectively with a variety of neurosurgical approaches. Often seizures can be controlled completely or at least dramatically improved with a concomitant improvement in the patient’s quality of life.[3] Surgical options include the transcallosal approach, pterional approach, orbitozygomatic approach, and transventricular endoscopic approach.
In our experience, the transcallosal approach appears to offer the best chance of controlling seizures even when resection of the hypothalamic hamartoma is incomplete. We believe that the improved outcomes associated with transcallosal resection (approaching the lesion from above) compared to procedures that approach and resect the hypothalamic hamartoma from below reflect more effective disconnection of the lesion as well as bulk resection. This explanation appears to be plausible. However, the pathway or pathways connecting the hypothalamic hamartoma tissue to other brain regions (pathways of seizure propagation) are uncertain. Physical proximity to limbic circuitry, including the columns of the fornix, mamillary bodies, and mammillothalamic tracts, makes propagation through limbic structures a potential candidate path.
Conclusion
Hypothalamic hamartomas offer a challenging diagnostic and management problem. Our current understanding of the pathophysiology of this disorder as it relates to seizure activity stresses both the intrinsic epileptogenesis of the hamartoma itself and its secondary effect on cortical function over time. Seizure activity arising from either the hamartoma or cortical regions appears to improve after resection of the hypothalamic hamartoma. Our experience with this lesion also raises questions about the utility of scalp video-EEG seizure monitoring for most hypothalamic hamartoma patients. Surgical resection of the hypothalamic hamartoma improves or potentially completely controls seizures either immediately or after a “running down” process of the secondary epileptogenesis involving cortical structures. If surgical resection of the lesion is incomplete, the success of seizure control may partially depend on the disruption of connections between the hamartoma and other brain regions.
References
- Arita K, Ikawa F, Kurisu K, et al: The relationship between magnetic resonance imaging findings and clinical manifestations of hypothalamic hamartoma. J Neurosurg 91:212-220, 1999
- Berkovic SF, Andermann F, Melanson D, et al: Hypothalamic hamartomas and ictal laughter: Evolution of a characteristic epileptic syndrome and diagnostic value of magnetic resonance imaging. Ann Neurol 23:429-439, 1988
- Berkovic SF, Arzimanoglou A, Kuzniecky R, et al: Hypothalamic hamartoma and seizures: A treatable epileptic encephalopathy. Epilepsia 44:969-973, 2003
- Berkovic SF, Kuzniecky RI, Andermann F: Human epileptogenesis and hypothalamic hamartomas: New lessons from an experiment of nature. Epilepsia 38:1-3, 1997
- Cascino GD, Andermann F, Berkovic SF, et al: Gelastic seizures and hypothalamic hamartomas: Evaluation of patients undergoing chronic intracranial EEG monitoring and outcome of surgical treatment. Neurology 43:747-750, 1993
- Daly DD, Mulder DW: Gelastic epilepsy. Neurology 7:189-192, 1957
- Diebler C, Ponsot G: Hamartomas of the tuber cinereum. Neuroradiology 25:93-101, 1983
- Dunoyer C, Ragheb J, Resnick T, et al: The use of stereotactic radiosurgery to treat intractable childhood partial epilepsy. Epilepsia 43:292-300, 2002
- Freeman JL, Harvey AS, Rosenfeld JV, et al: Generalized epilepsy in hypothalamic hamartoma: Evolution and postoperative resolution. Neurology 60:762-767, 2003
- Fukuda M, Kameyama S, Wachi M, et al: Stereotaxy for hypothalamic hamartoma with intractable gelastic seizures: Technical case report. Neurosurgery 44:1347-1350, 1999
- Kahane P, Munari C, Minotti L, et al: The role of the hypothalamic hamartoma in the genesis of gelastic and dacrystic seizures, in Tuxhorn I, Hothausen H, Boenigk H (eds): Paediatric Epilepsy Syndromes and Their Surgical Treatment. London: John Libbey, 1997, pp 447-461
- Kuzniecky R, Guthrie B, Mountz J, et al: Intrinsic epileptogenesis of hypothalamic hamartomas in gelastic epilepsy. Ann Neurol 42:60-67, 1997
- Munari C, Kahane P, Francione S, et al: Role of the hypothalamic hamartoma in the genesis of gelastic fits (a video-stereo-EEG study). Electroencephalogr Clin Neurophysiol 95:154-160, 1995
- Murphy JV, Wheless JW, Schmoll CM: Left vagal nerve stimulation in six patients with hypothalamic hamartomas. Pediatr Neurol 23: 167-168, 2000
- Palmini A, Chandler C, Andermann F, et al: Resection of the lesion in patients with hypothalamic hamartomas and catastrophic epilepsy. Neurology 58:1338-1347, 2002
- Parrent AG: Stereotactic radiofrequency ablation for the treatment of gelastic seizures associated with hypothalamic hamartoma. Case report. J Neurosurg 91:881-884, 1999
- Regis J, Bartolomei F, de Toffol B, et al: Gamma knife surgery for epilepsy related to hypothalamic hamartomas. Neurosurgery 47:1343-1351, 2000
- Regis J, Bartolomei F, Hayashi M, et al: The role of Gamma knife surgery in the treatment of severe epilepsies. Epileptic Disord 2:113-122, 2000
- Rosenfeld JV, Harvey AS, Wrennall J, et al: Transcallosal resection of hypothalamic hamartomas, with control of seizures, in children with gelastic epilepsy. Neurosurgery 48:108-118, 2001
- Zaatreh M, Tennison M, Greenwood RS: Successful treatment of hypothalamic seizures and precocious puberty with GnRH analogue. Neurology 55:1908-1910, 2000