
Lateral Ventricle Neoplasm with Skeletal Muscle Differentiation
Andrew Little, MD
Jonathan S. Hott, MD*
Kris A. Smith, MD
Stephen W. Coons, MD**
Divisions of Neurological Surgery and **Neuropathology, Barrow Neurological Institute, St. Joseph’s Hospital and Medical Center, Phoenix, Arizona *Current Address: John C. Lincoln Health Network, Phoenix, Arizona
Abstract
This case report describes the clinical, imaging, surgical, and pathological findings of an intraventricular neoplasm with skeletal muscle features in a 15-year-old girl. The lesion occupied the right lateral ventricle and enhanced intensely on T1-weighted MR images. Intraoperatively, the lesion appeared to derive its blood supply from the choroid plexus. Because the neoplasm lacked parenchymal invasion and was contained within a dense capsule, it was easily resected en bloc. Microscopically, the lesion had mixed rhabdoid and epithelioid features and a low-grade phenotype. This intraventricular neoplasm with skeletal muscle differentiation may represent a previously unreported tumor. Theories about its origin are discussed, and the implications of the lesion’s unique characteristics on treatment are reviewed.
Key Words: choroid plexus papilloma, desmin, intraventricular neoplasm, rhabdomyosarcoma, skeletal muscle
Abbreviations Used: MR, magnetic resonance
Primary intracranial tumors with skeletal muscle differentiation are rare. Such lesions include teratomas, medullomyoblastomas, and rhabdomyosarcomas.[1,3,4,6,7,10,13,16,17] Together, they occur in a number of locations, including the sella, posterior fossa, cortical parenchyma, and within the ventricular system. Intraventricular tumors represent 10% of intracranial neoplasms. In older children and young adults choroid plexus papillomas, meningiomas, ependymomas, and glial tumors are among the most common neoplasms in the lateral ventricles.[8,9,12,15] This report describes the case of a girl with a ventricular neoplasm, which had the clinical and radiological appearance of a choroid plexus papilloma. Histopathologically, it most closely resembled an embryonal rhabdomyosarcoma, but it appears to have been a distinct lesion with low-grade features.
Case Report
A 15-year-old Hispanic girl had headaches for several months and a syncopal episode. Her physical examination was normal. MR imaging of the brain showed a well-demarcated, 1.9 x 2.0-cm, enhancing lesion in the atria of the right lateral ventricle (Fig. 1). The lesion was associated with the choroid plexus and did not invade the brain parenchyma. There was no hydrocephalus. Given the patient’s age and imaging findings, choroid plexus papilloma was considered to be the most likely diagnosis.
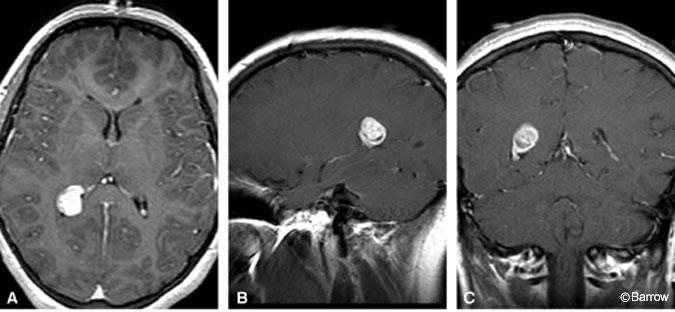
The patient underwent a right parieto-occipital craniotomy with stereotactic guidance. Intraoperatively, a discrete mass derived its blood supply from the choroid plexus, raising the possibility that it was a choroid plexus papilloma. The lesion was removed as a single specimen. Grossly, it was solid, soft, and pink.
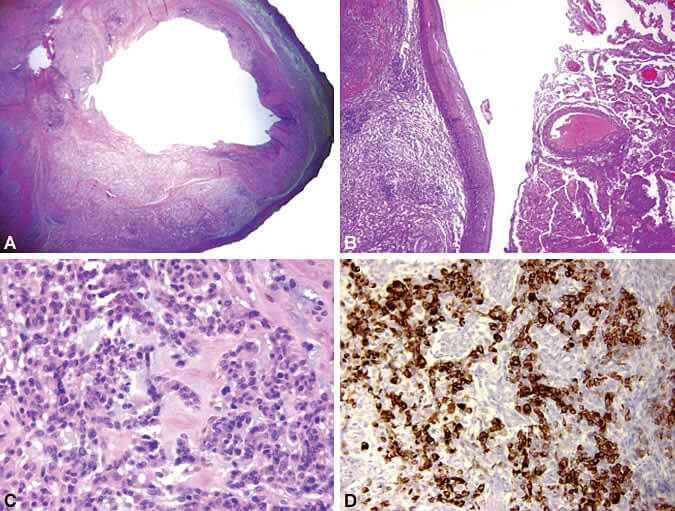
Histopathological analysis demonstrated a cystic lesion with a dense fibrogliotic wall (Fig. 2A). The neoplasm was attached to, but distinct from, normal choroid plexus (Fig. 2B). Tumor cells were arranged in lobules and cords within a fibromyxoid stroma. The cells had mixed epithelioid, spindle, and rhabdoid morphologies. They contained oval nuclei with brightly eosinophilic cytoplasm (Fig. 2C). They were strongly reactive for desmin (Fig. 2D) but negative for S-100, epithelial membrane antigen, synaptophysin, neurofilament, keratin CAM 5.2, and smooth muscle-and muscle-specific actin. Activity to glial fibrillary acidic protein was patchy. Interestingly, several low-grade features were noted: occasional mitotic figures, MIB-1 labeling index of 1 to 3%, and scant atypia. Analysis confirmed that the lesion did not invade the adjacent parenchyma. Together, these findings were considered inconsistent with other intracranial neoplasms such as rhabdomyosarcomas. Although gliomas may exhibit desmin reactivity, this tumor appears to be a distinct entity. Our findings were verified by three neuropathologists at another institution.
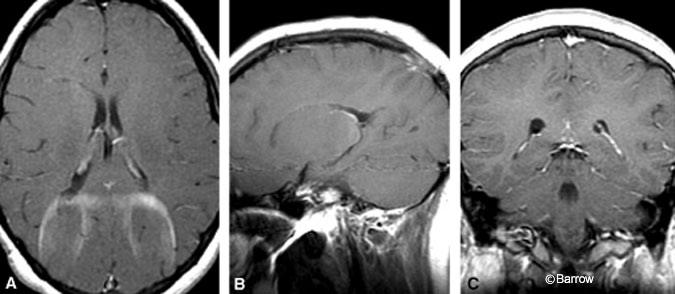
Postoperative MR imaging confirmed complete resection of the mass (Fig. 3). Eighteen months after surgery, the patient had no evidence of recurrence on surveillance imaging. Her headaches have resolved, and she has had no further syncopal episodes. She has received no further treatment.
Discussion
Primary intracranial neoplasms with skeletal muscle differentiation are rare. Several reports have described lesions in the ventricles, parenchyma, and sellar region.[1,3,4,6,7,13,16] At presentation, these tumors are often large. Their clinical course is aggressive, and they are difficult to resect completely. Their imaging characteristics vary, but they tend to be iso- to hyperdense on T1-weighted MR images and to demonstrate patchy enhancement. Imaging findings such as the presence of necrotic foci and surrounding brain edema are consistent with their high-grade nature.[5] In contrast, this patient’s lesion enhanced intensely, was well marginated, and lacked surrounding edema. Furthermore, the lesion was surrounded by a dense capsule and was easily removed.
Microscopically, intracranial rhabdomyosarcomas have features identical to those of rhabdomyosarcomas originating elsewhere. They are characterized by rhabdomyoblasts of various shapes, densely arrayed cells within a myxoid stroma, atypia, a high mitotic index, and the expression of markers of skeletal muscle differentiation such as desmin.[14] Our patient’s lesion shared some of these characteristics, including the presence of rhabdomyoblasts (although the degree of atypia was mild), myxoid stroma, and desmin positivity. However, it contained a prominent capsule and had low mitotic and MIB-1 labeling indices. That our patient had no evidence of a recurrence 18 months after resection further supports its low-grade nature.
Teratomas may also contain skeletal muscle components.[4] True teratomas contain elements of all three germ layers and often occupy a midline location in the central nervous system. Our patient’s lesion was lateral to midline and contained only connective tissue components.
Finally, medullomyoblastomas, which classically arise in the posterior fossa, may exhibit mesenchymal characteristics.[10,11] Unlike medulloblastomas, this neoplasm had cells that were larger than those seen in primitive neuroectodermal tumors. Furthermore, it exhibited no glial or neuronal differentiation and contained a myxoid stroma.
The histogenesis of this tumor is unknown. Others have speculated that mesenchymal tumors of the brain originate from pericapillary cells derived from the mesoderm or from the pluripotent cells of the neural crest.[4] Interestingly, this lesion appeared to derive its blood supply from the choroid plexus, a finding that may have implications for its origin. The mesenchyma of the choroid plexus develops from the meninges, which in turn develops from the neural crest.[2] Therefore, we speculate that the lesion may have originated from neural crest cells.
The optimal treatment for this patient has not been defined. Given that complete resection seemed to have been achieved and that the pathology of the tumor was low grade, we elected to follow the patient rather than to proceed with adjuvant therapy as would be standard for embryonal rhabdomyosarcomas. We believe that this case raises an important question regarding applying standard treatment regimens to novel tumors. In this instance we let the unique aspects of the case guide therapy. The observation that this girl was disease free 18 months after resection supports this strategy.
This intraventricular neoplasm with rhabdoid characteristics was similar to an embryonal rhabdomyosarcoma but appears to have been a distinct tumor. Given its unique attributes, we believe that the tumor may represent a previously unreported and unclassified low-grade neoplasm with skeletal muscle features.
Acknowledgment
We thank Dr. Gregory Fuller for his assistance in the pathological evaluation of the tumor.
References
- Arita K, Sugiyama K, Tominaga A, et al: Intrasellar rhabdomyosarcoma: Case report. Neurosurgery 48:677-680, 2001
- Catala M: Embryonic and fetal development of structures associated with the cerebro-spinal fluid in man and other species. Part I: The ventricular system, meninges and choroid plexuses. Arch Anat Cytol Pathol 46:153-169, 1998
- Celli P, Cervoni L, Maraglino C: Primary rhabdomyosarcoma of the brain: Observations on a case with clinical and radiological evidence of cure. J Neurooncol 36:259-267, 1998
- Dropcho EJ, Allen JC: Primary intracranial rhabdomyosarcoma: Case report and review of the literature. J Neurooncol 5:139-150, 1987
- Evans A, Ganatra R, Morris SJ: Imaging features of primary malignant rhabdoid tumour of the brain. Pediatr Radiol 31:631-633, 2001
- Hanna SL, Langston JW, Parham DM, et al: Primary malignant rhabdoid tumor of the brain: clinical, imaging, and pathologic findings. AJNR Am J Neuroradiol 14:107-115, 1993
- Herva R, Serlo W, Laitinen J, et al: Intraventricular rhabdomyosarcoma after resection of hyperplastic choroid plexus. Acta Neuropathol (Berl) 92:213-216, 1996
- Jelinek J, Smirniotopoulos JG, Parisi JE, et al: Lateral ventricular neoplasms of the brain: Differential diagnosis based on clinical, CT, and MR findings. AJNR Am J Neuroradiol 11:567-574, 1990
- Kendall B, Reider-Grosswasser I, Valentine A: Diagnosis of masses presenting within the ventricles on computed tomography. Neuroradiology 25:11-22, 1983
- Lewis AJ: Medulloblastoma with striated muscle fibers. Case report. J Neurosurg 38:642-646, 1973
- Misugi K, Liss L: Medulloblastoma with cross-striated muscle. A fine structural study. Cancer 25:1279-1285, 1970
- Morrison G, Sobel DF, Kelley WM, et al: Intraventricular mass lesions. Radiology 153:435-442, 1984
- Preissig SH, Smith MT, Huntington HW: Rhabdomyosarcoma arising in a pineal teratoma. Cancer 44:281-284, 1979
- Qualman SJ, Coffin CM, Newton WA, et al: Intergroup Rhabdomyosarcoma Study: Update for pathologists. Pediatr Dev Pathol 1:550-561, 1998
- Silver AJ, Ganti SR, Hilal SK: Computed tomography of tumors involving the atria of the lateral ventricles. Radiology 145:71-78, 1982
- Tomei G, Grimoldi N, Cappricci E, et al: Primary intracranial rhabdomyosarcoma: Report of two cases. Childs Nerv Syst 5:246-249, 1989
- Wright DH, Naul LG, Hise JH, et al: Intraventricular fibroma: MR and pathologic comparison. AJNR Am J Neuroradiol 14:491-492, 1993