
Molecular Biology of Chemotherapy and Resistance
Adrienne C. Scheck, PhD
Division of Neurology, Barrow Neurological Institute, St. Joseph’s Hospital and Medical Center, Phoenix, Arizona
Abstract
The recurrence of human gliomas several months after treatment suggests that intrinsic, rather than acquired, resistance underlies the failure of therapy. Therapy reduces cellular heterogeneity, allowing only resistant cells to survive. Mechanisms of therapy resistance must be understood before new treatment modalities can be developed. Consequently, this article reviews resistance to therapy-induced cell death after treatment with a variety of chemotherapeutic alkylating agents and topoisomerase inhibitors, along with the role of gene therapy in designing more efficacious< therapies.
Key Words : apoptosis, BCNU, cisplatin, chemotherapy, gene therapy, gliomas, resistance, tumors
Human malignant glioma is an almost uniformly fatal disease. Despite decades of research, life expectancy after its diagnosis has not substantially changed. Surgical resection of the primary tumor followed by irradiation and chemotherapy is a palliative measure, and the tumor typically recurs. The recurrent tumor is often detected 3 or 4 months after treatment, suggesting that intrinsic rather than acquired resistance is responsible for the failure of therapy. Thus, within the heterogeneous population of cells that comprises the primary tumor, a subpopulation of cells is intrinsically resistant to therapy and capable of surviving and growing in the tumor bed after therapy.57, 91 If the mechanisms of therapy resistance could be determined, the way would be paved for the development and application of additional adjuvant therapies and treatment modalities.
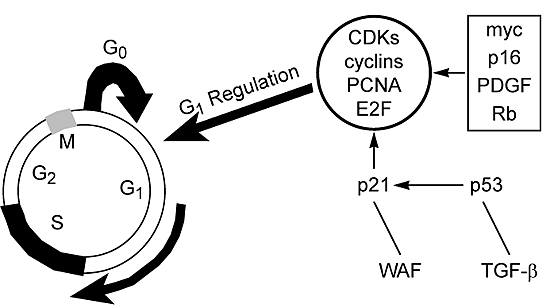
The Cell Cycle, Apoptosis, and Resistance to Therapy
Considerable interest has been focused on the cell cycle and the role of the tumor suppressor p53 in investigations of resistance to therapy. During normal cell growth and division, the cell traverses through the cell cycle in an ordered manner (Fig. 1). At the boundaries of G1®S and G2®M, however, there are “checkpoints” where the cell cycle can stop if damage to the deoxyribonucleic acid (DNA) is detected. At these times the cell may attempt to repair the damage, and a decision is made either to continue the cell cycle or to initiate a cascade of events that leads to cell death through a process known as programmed cell death or apoptosis. A complete discussion of apoptosis is outside the scope of this review; however, the importance of this process in the cellular response to therapy warrants a brief discussion.
Apoptosis is characterized by membrane blebbing with intact membranes, cell shrinkage leading to the formation of apoptotic bodies, chromatin condensation, and nonrandom DNA fragmentation that causes DNA laddering. The “balance” of a number of genes, including the pro-apoptotic genes bax, bak and bad as well as the apoptosis suppressors bcl-2 and bclxL, affects cell survival or apoptosis directly. In many systems, apoptosis is initiated rapidly after cytotoxic therapy, and the process is often dose dependent.
Resistance to therapy-induced apoptosis may be a mechanism of resistance in a variety of tumor systems and is likely to be linked to alterations in the cell cycle at the checkpoints as a result of therapy-induced DNA damage. Thus, the regulation of the cell cycle at these checkpoints is crucial to how cells respond to therapy. Correspondingly, the roles of TP53 and related genes [e.g., p21WAF1/CIP1and murine double minute 2 oncogene (MDM2)] in this response have received considerable attention.38
In some cells with a wild type p53 phenotype, p53 is induced by damage to the DNA. Its induction causes the arrest of G1, which allows time for the DNA to be repaired before its replication. If the DNA is repaired, the cell goes through a normal cell cycle. If the DNA cannot be repaired, cells may undergo apoptosis,29 presumably through the up-regulation of bax and the down-regulation of bcl-2 expression. The cell cycle of cells with a mutated or missing TP53 gene may not be arrested.47,66 Consequently, the damaged DNA is replicated and may contribute to tumor progression.
Alterations in genes that modulate the activity of p53 can mimic the p53-deficient phenotype, even in the presence of a normal TP53 gene. For example, the p21WAF1/CIP1 gene is induced by p53 and may be responsible for the p53-dependent checkpoint at G1. Colorectal cancer cells deficient in p21 do not undergo p53-dependent arrest of G1 after DNA damage,112 either in vivo or in vitro.24Additional studies have demonstrated that p21-deficient cells undergo apoptosis after DNA damage.113 In some systems, however, p53 is required for the initiation of apoptosis.56,66 DNA damage caused by radiation or chemotherapeutic agents such as cis-diaminedichloroplatinum(II) (DDP, cisplatin) can increase the levels of messenger ribonucleic acid (mRNA) encoding MDM2.80Furthermore, the accumulation of p53 protein increases in cisplatin-resistant ovarian tumor cell lines.12,29 Adenovirus- or transfection-mediated transfer of wild-type p53 into cells increases their sensitivity to cisplatin.12,31 In contrast, astrocytes from p53 knockout mice with wild-type p53 were more resistant to 1,3-bis-(2-chlorethyl)-1-nitrosourea (BCNU, carmustine) than those from p53-null mice.71Thus, the action or actions of TP53 and related genes with respect to arrest of the cell cycle, apoptosis, and/or resistance to therapy vary in different cell types and depend on the genetic background of the cells (e.g., other oncogenes, growth factors). An understanding of the full impact of these genes on resistance to therapy is likely to require additional insights into the inter-relationships between these genes and other modulators of cell growth.
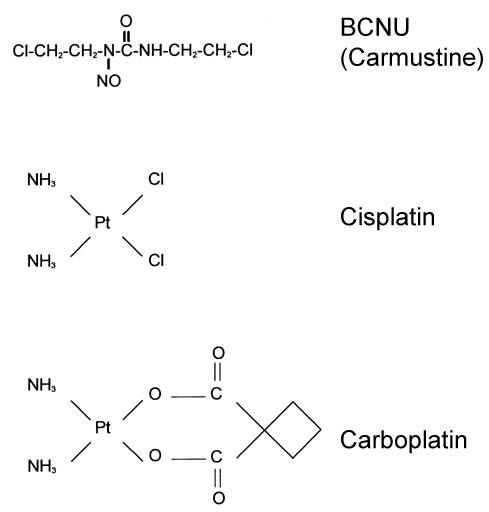
Alkylating Agents
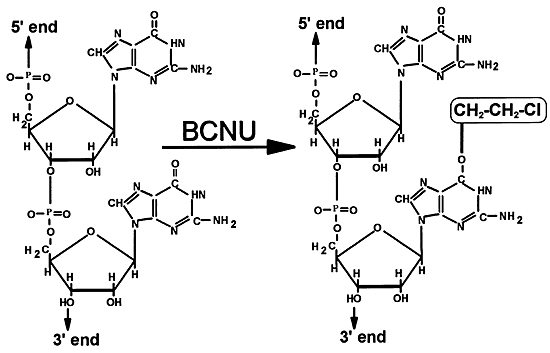
The chemotherapeutic agent most commonly used in the treatment of gliomas is BCNU (Fig. 2), a lipophilic, bifunctional, alkylating agent that crosses the blood-brain barrier. This feature facilitates its use in brain tumor therapy. In an aqueous environment, BCNU acts as both an alkylating and a carbamoylating agent capable of damaging both nucleic acids and proteins. The major cytotoxic event related to BCNU treatment is alkylation of guanine at the O6 position (Fig. 3). If unrepaired, this damage leads to misreading of the DNA code, to covalent cross-linking of the DNA, or both.
In addition to BCNU, similar agents such as cisplatin and its analogue cis-diamine 1,1-cyclobutane dicarboxylatoplatinum II (carboplatin) are used in the treatment of gliomas (Fig. 2). Like BCNU, these drugs also form intra- and interstrand cross-links in DNA and protein. Streptozotocin is an antibiotic isolated from Streptomyces achromogenes . Its principal mechanism of action is thought to be methylation of DNA; it does not alkylate DNA like BCNU.
A number of genes may be involved in resistance to BCNU and other alkylating agents. For example, the gene for methylguanine methyltransferase (MGMT) is located on chromosome 10,83 and MGMT is active in the repair of nitrosourea-induced DNA damage. This protein reacts with the DNA such that the alkyl group is transferred from the DNA molecule to a cysteine acceptor site. The acceptor site cannot be regenerated. Thus, MGMT is known as a suicide enzyme—each protein molecule can remove one alkyl group from the DNA and is then irreversibly inactivated. Typically, cells with little or no methyltransferase activity (often termed mer-) are more sensitive to alkylating agents than are cells that express this activity (termed mer+). MGMT is expressed in human brain tumors30,102,103 and is elevated in some but not all BCNU-resistant glioma cell lines.76,86 Furthermore, the expression of this protein is heterogenous within cells in the same tumor,52 and alkyltransferase immunoreactivity may be correlated with the survival of patients treated with BCNU.7
The gene encoding MGMT has been cloned.40,108,111 Furthermore, the expression of MGMT with concomitant alterations in sensitivity to BCNU can be modulated through transfection experiments (to increase MGMT activity).23 Compounds such as O6-benzylguanine and streptozotocin inactivate MGMT and increase the cytotoxicity of BCNU in resistant cells.2,20,34-36,58-60,78 In addition to MGMT-mediated repair, DNA mismatch repair22 and nucleotide excision repair14,15 (Scheck and Shah, unpublished data) may be involved in resistance to BCNU. Thus, the role of DNA repair in resistance to alkylating agents is likely to be multifaceted, and intervention may therefore require multiple approaches.
Cells may also be resistant to alkylating agents through detoxification via conjugation with glutathione (GSH). GSH is an important component of drug resistance in gliomas, and the overexpression of enzymes involved in the GSH redox cycle [e.g., glutathione-S-transferases (GSTs) and gammaglutamyl transpeptidase (GGT)] has been implicated in resistance to alkylating agents. The prevention of DNA damage by the “scavenging” of free radicals by sulfhydryl compounds such as GSH may also contribute to intrinsic radioresistance.9,18,63 Reduction of the amount of GSH with drugs such as buthionine sulfoximine increases a cell’s sensitivity to both radiation and chemotherapy.73 Three classes of GST isoenzymes (a, m, p) have been implicated in resistance to alkylating agents in a number of human and animal cells, including gliomas.17,51,81,101,105,109,114 In glioma cell lines, the expression of GST-p also correlates with the degree of resistance to BCNU.3
[one_half]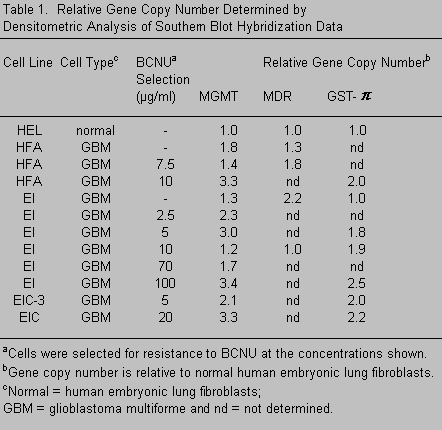
[/one_half]
[one_half_last]
[/one_half_last]
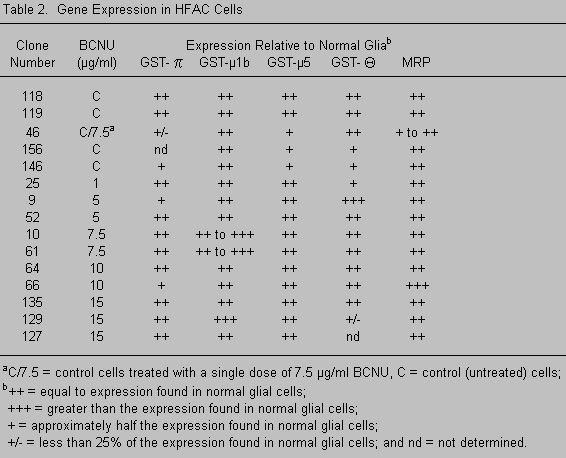
Another enzyme in the GSH cycle, GGT, is involved in the biosynthesis of GSH. GGT is mapped to chromosome 22q11.2 and may play a role in the resistance of some human malignant glioma cell lines106 and other tumor cells53 to nitrosoureas. Furthermore, the multidrug resistance-associated protein (MRP) has been implicated in the GST-mediated detoxification by acting as an efflux pump for GSH-conjugated molecules.67,118 However, the gene copy number and/or expression of these genes is not elevated in all resistant cells (Tables 1 and 2). The multidrug resistance gene (MDR) locus is mapped to chromosome 7 and encodes a highly homologous family of proteins that also act as energy-dependent drug efflux pumps.25 The presence of p-glycoprotein (the MDR gene product) has been found only in the endothelial cells of normal brain,19 and the expression of protein, mRNA, or both varies in some, but not all, gliomas and/or glioma cell lines.6,41,61,68 We, too, have found only a small increase in the gene copy number and mRNA expression of the MDR gene in only a few glioma cell lines (Table 1). Overexpression of this protein has not been demonstrated in BCNU-resistant gliomas,68 and cells with an MDR phenotype are often, but not always, sensitive to BCNU and cisplatin.44,45
A number of resistance mechanisms have been described for cisplatin and other platinum-containing drugs, mostly from work with ovarian cancer cells. A number of genes play roles similar to those involved in resistance to BCNU. GSH and genes of the GSH cycle may be elevated in resistant cells,4,39,48,51,62 leading to drug detoxification. Reduction of GSH by transfection of a GST-p antisense construct also reduces the resistance of a colon cancer cell line to cisplatin.4 Furthermore, the expression or activity of genes involved in DNA repair, such as those of the thymidylate synthase cycle84 and DNA polymerase b,49 increases in some cells.
Recently, attention has shifted to mechanisms of DNA repair, including gene interactions and mismatch repair.1,13,21,26,27,69,79,104The isolation of genetic suppressor elements and changes in sensitivity to cisplatin when these elements are introduced into resistant cells suggest a role for p53 in resistance to cisplatin.33 Furthermore, mutations of TP53 are found in cells resistant to cisplatin.75
Resistance to Alkylating Agents
Comparative genomic hybridization studies of two human ovarian cancer cell lines and derivatives selected for resistance to cisplatin have shown a gain of material from chromosomes 2q, 4, 6q and 8q as well as a loss of material from chromosomes X, 2p, 7p, 11p, and 13.115 In contrast, somatic cell hybridization studies using one of the same ovarian carcinoma cell lines have demonstrated that resistance segregated with chromosomes 11 and 16. Whether a gene or genes from these chromosomes are actively involved in resistance to cisplatin remains to be seen. Such studies, however, provide a background on which to base further molecular studies. This is the strategy used in our laboratory to analyze BCNU resistance in gliomas.
Although many brain tumor cells are sensitive to damage by BCNU, the karyotypic heterogeneity in primary malignant gliomas100 and differential sensitivity of individual cell types to BCNU are substantial.117 Bradford et al.10 replicated these findings and also showed differences in the drug sensitivity of clones derived from a murine astrocytoma. At the level of individual genes,86,88,89 human glioma cell lines show a similar heterogeneity.
When cells from the primary tumor are grown in the presence of clinically achievable concentrations of BCNU (blood plasma levels, ~10 mg/ml) or are cloned from a colony-forming assay after a single exposure to BCNU,97 a BCNU-resistant subpopulation of cells is selected in vitro that is near-diploid.97 These cells carry a specific nonrandom karyotypic deviation that includes overrepresentation of fragments or whole chromosomes 7 and 22.87,92 Approximately 30% of the tumors studied to date have this subpopulation at the time of the primary resection. When treated with radiation and BCNU, these patients develop recurrent tumors in which this same cell type (near-diploid with overrepresentation of some or all of chromosomes 7 and 22) is the dominant population.99 Our molecular analyses of drug-resistant cells with overrepresentation of chromosomes 7 and 22 have not consistently detected large differences in the copy number or expression of genes known to be involved in BCNU resistance (Tables 1 and 2).
A new class of GST—GST-q—has been localized to chromosome 22. There are at least two isoenzymes in this family,43 one of which is polymorphic and absent from approximately 38% of the population.74
GST-q appears to play a role in detoxifying carcinogenic chemicals such as halomethanes;74 however, a role for this enzyme in resistance to chemotherapy has not yet been described.
To determine if this isoenzyme is involved in resistance to BCNU, we used a cell line that lacked MGMT activity. BCNU-resistant clones were isolated from this cell line, and the expression of GST isoenzymes was determined (Table 2). GST-q did not appear to play a role in BCNU resistance in these cells. Other GST isoenzymes were overexpressed in some, but not all, resistant clones (Table 2). Furthermore, genes known to be involved in BCNU resistance (mainly MGMT and the GSTs) are not mapped to chromosomes 7 and 22 (except GST-q). This finding suggests that the overrepresentation of chromosomes 7 and 22 involves other genes related to survival, growth, or both after drug therapy.
Nor is drug resistance a simple matter of increased gene copy number resulting from karyotypic abnormalities. MGMT is mapped to chromosome 10, a chromosome frequently underrepresented in gliomas. Many of our cell lines, however, contain a normal or increased copy number and expression of the MGMT gene. So, perhaps, while the chromosome is lost, this gene (which is probably required for survival in some cells after drug treatment) is functionally retained somewhere in the genome. This possibility underscores the importance of gene expression studies. We have also correlated overexpression of the GSTs with BCNU resistance in some, but not all, of our BCNU-resistant tumor cell lines (Table 2).
The lack of consistent correlation between the expression of MGMT and/or GSTs and drug resistance has a number of possible explanations. It is likely that gliomas use more than one mechanism to survive adjuvant therapies. Our work and most of that reported in the literature have demonstrated that both MGMT and a number of GST isoforms are involved in BCNU resistance. The heterogeneity that is prevalent in these tumors extends to drug-resistance mechanisms. Although some tumors appear to use GST(s) or MGMT, some appear to use both and some do not appear to use either. Liang55 reached a similar conclusion when he was unable to demonstrate increases in the expression of these same genes after hypoxia-induced increases in BCNU- and cisplatin-resistance in human glioma cell lines.
Topoisomerase II Inhibitors: Etoposide and Teniposide
The topoisomerases play a crucial role in the normal processes of DNA replication and transcription by facilitating DNA breakage that allows DNA supercoils to relax. Topoisomerase II specifically cleaves double-stranded DNA and bridges the break while another piece of double-stranded DNA passes through. Etoposide (VP-16) and teniposide (VM-26) are topoisomerase II inhibitors that exert their effect by stabilizing the cleavable complex of the DNA-topoisomerase II. These drugs do not directly interact with DNA, suggesting a mechanism of action (and mechanism(s) of resistance) specifically related to the topoisomerase II enzyme.
Resistance to these drugs could result from diminished amounts of topoisomerase II. In general, quiescent cells have little detectable topoisomerase II. In contrast, proliferating cells have many copies of these molecules, suggesting that faster-growing tumors should be more sensitive to these therapies than tumors that grow more slowly. However, topoisomerase II is down-regulated in some resistant tumors. Considerable evidence indicates that alterations in one or more domains of the topoisomerase II molecule itself can create resistance. Molecules with increased resistance to a cleavable-complex formation, altered catalytic activity, an altered ATP-binding domain, and/or an altered drug-binding domain can affect the cells’ sensitivity to VP-16 and VM-26.37 Furthermore, although the cleavable complex is relatively short lived, the presence of these complexes leads to permanent DNA damage. This damage is likely mediated by the DNA synthesis machinery itself because inhibitors of DNA synthesis can (at least partially) restore resistance.
Gene Therapy
A number of approaches to gene therapy have been used in attempts to design more efficacious therapies and are reviewed elsewhere.82 Briefly, however, these therapies are composed of two main “parts”: a vector-delivery system and a therapeutic agent. The vector-delivery system can be a genetically engineered virus, plasmid, or cell that is somehow delivered or targeted to the tumor. Many of these proposed therapies have shown promise in rodent models. Their efficacy in humans, however, is under intense scrutiny and has yet to be established.
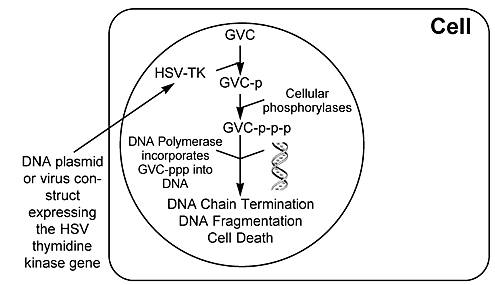
Perhaps the best known therapy is the HSV-tk/ganciclovir system (Fig. 4). In this strategy, the thymidine kinase (tk) gene from theHerpes simplex virus (HSV) is delivered to the tumor cell using a virus vector such as a genetically altered adeno- or retrovirus. The patient is then treated with ganciclovir. The HSV-tk phosphorylates the ganciclovir to the monophosphate form. Cellular phosphorylases can then phosphorylate it to the triphosphate form. In this form it is incorporated into cellular DNA, resulting in chain termination causing cell death. Despite its successful use in rodent models, this therapy has met with limited success in humans.
Ongoing trials with this therapy, however, are providing information about the cytotoxic effects of this system on cells with and without the virus construct. The so-called “bystander effect” appears to mediate cytotoxicity in cells that have not received the HSV-tk gene. The mechanisms underlying this effect are unknown, but they may be related to the transfer of metabolic products through gap junctions, phagocytosis of dead or dying cells, or changes in the host’s immune system. Regardless of its source, the bystander effect is an important part of gene therapy.
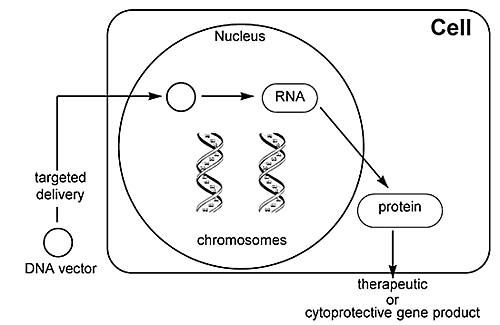
The transfer of drug-resistant genes (Fig. 5) such as MGMT or the multidrug resistant gene (MDR-1) is also being studied in an attempt to “protect” the host from chemotherapeutic side effects, thereby allowing the use of higher doses of toxic therapeutic agents. Thus, the addition of MGMT to hematopoietic cells helps to protect these cells from the unwanted side effects of treatment with BCNU. Genes that modulate the host’s immune system are also being transduced in an attempt to cause the host to mount an immune response against the tumor.
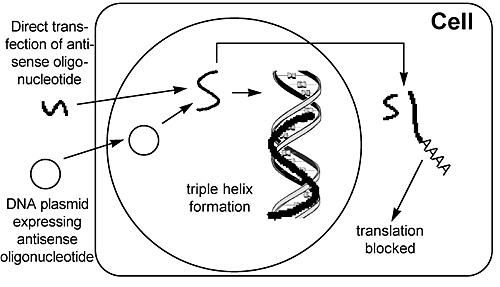
In addition to therapies directly involving chemotherapeutic agents, trials in which oncogene expression is reduced or blocked are ongoing. In these systems, a vector is used to deliver an antisense molecule (Fig. 6) or a ribozyme (Fig. 7) specific for a particular oncogene. Antisense molecules are nucleic acid fragments whose sequence is complementary to the gene of interest. When added to the cell, it base-pairs with the mRNA or DNA and inhibits transcription, splicing, transport, and/or translation of the message. Ribozymes are catalytic RNA molecules designed to cut a specific sequence in the mRNA molecule. In both cases, the expression of the targeted oncogene is reduced.
Tumor suppressor genes are another avenue of investigation. Some gene therapies, for example, are designed to reintroduce wild type tumor suppressor genes (such as TP53) to cells lacking its normal expression. Also being tested are strategies that block or inhibit a tumor’s ability to induce angiogenesis, thus preventing the tumor from enhancing its blood supply.
Investigations of new therapies are ongoing.16 More than 200 new agents are being tested in clinical trials of all forms of cancer. Despite the enormous increase in gene therapy trials during the last few years, a major hurdle must be overcome before these therapies can be used to their full potential. The delivery systems used to deliver these genes are still far from optimal. Vectors and delivery systems must be designed to deliver the genes safely to the appropriate cells and to express them in a therapeutically useful way.
Molecular Biology and the Future of Brain Tumor Therapy
The presence of intrinsically resistant cells in the absence of the overexpression of a particular gene or genes suggests the presence of other mechanism(s) of resistance. A possible explanation for such resistance may relate to gene interactions that occur in response to therapy. One such example is the postulated connection between immediate-early regulatory genes and resistance to therapy. In particular, work in a number of systems has demonstrated a link between the expression of c-fos and the induction of genes involved in DNA repair and/or drug resistance.
The expression of c-fos can be altered by treatment with alkylating agents. These changes have been demonstrated in two cancer cell lines. In a colon carcinoma line, the steady-state levels of c-myc and c-fos transcripts have been increased.32 In a cisplatin-resistant ovarian carcinoma line, the expression of c-fos was increased along with c-H-ras, thymidylate synthetase, and DNA polymerase b.46Suppressing the induction of c-fos reduces these cells’ resistance to drugs.85 The induction of GST is mediated by regulatory elements that are activated by fos/jun heterodimeric complexes (AP-1).8 Therefore, the induction of c-fos by a growth factor or other mechanism may stimulate the production of genes associated with DNA repair, resulting in a more resistant phenotype. In addition to gene interactions that alter transcriptional activation, genes involved in the control of the cell cycle may play a role in DNA repair and resistance to therapy, as described above.
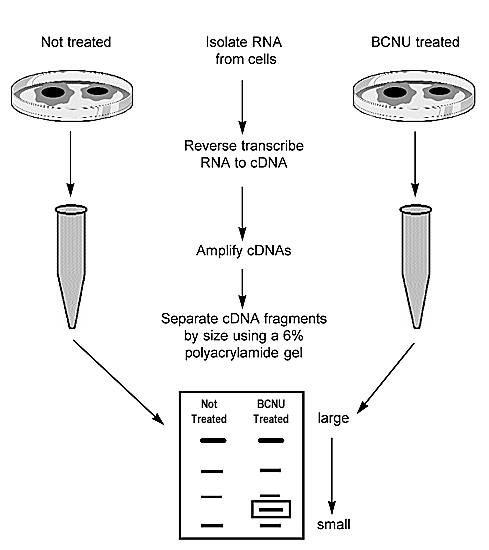
Thus, the role of many genes and gene interactions in the resistance to therapy is only now being realized. Likely, other as yet unidentified genes are also involved. Our work is aimed at identifying these genes by comparing gene expression in drug-sensitive cells with that of drug-resistant cells using a technique known as differential mRNA display (Fig. 8).
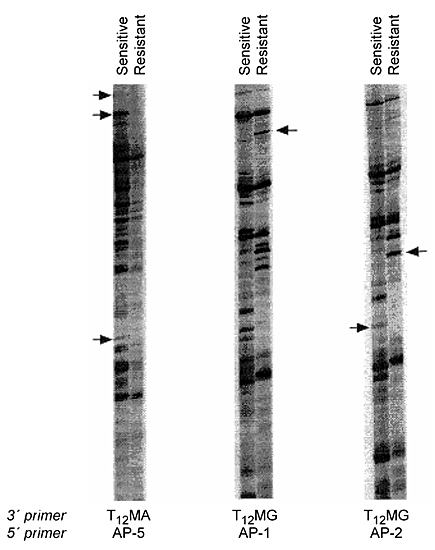
This technique is based on reverse transcription-polymerase chain reaction technology. Instead of designing primers to a specific gene, however, random primers are used so that all mRNAs in the cell are amplified. These RNAs are then separated and visualized on a polyacrylamide gel, and the resulting patterns (representing gene fragments) are compared (Fig. 9). Differences in the pattern may be indicative of differential gene expression. These fragments are then cloned and sequenced to determine their identity.
We have used this strategy to identify genes that may be involved in resistance to BCNU therapy. The presence of differentially expressed RNAs in BCNU-sensitive (untreated) and BCNU-resistant (5 µg/ml) cells was demonstrated,70,90 and the fragments were reamplified, cloned, and sequenced. We have identified one overexpressed gene as the transforming growth factor b1 binding protein (LTBP-1).
The potential role of LTBP-1 in BCNU resistance is completely unknown and has not been described previously. LTBP-1 is a component of the latent transforming growth factor b (TGF-b) complex that is required for activation of TGF-b1. The gene encoding this protein has been mapped to 19q13.1, and additional related proteins have recently been identified.28,64,107,110 The precise role of these proteins in the activation of latent TGF-b1 has not been determined, but the overexpression of this gene in BCNU-resistant HFA cells is intriguing. TGF-b1 interacts with platelet-derived growth factor (PDGF) and/or its receptors,5,11,50,54,72,116 and it may be involved in apoptosis65 or in resistance to the therapeutic agent N-phosphonacetyl-L-aspartate (PALA).42 Furthermore, overexpression of this gene could have an indirect effect on the regulation of the cell cycle, which may also ultimately affect the cell’s ability to survive therapy.
Given that some BCNU-resistant cells overexpress PDGF,87 this finding is particularly intriguing. The roles of this gene and other as yet unidentified genes found by our differential mRNA display experiments in BCNU resistance are being investigated. Through these studies we hope to identify novel mechanisms of therapy resistance.
Conclusions
The resistance mechanisms in tumors cannot be analyzed without understanding the biology of the system. Most techniques are “population” techniques—that is, the results represent an average of many cells. Even data obtained from cultured cells can be influenced by the dynamics of the culture and the selection pressure(s) placed on cells by their environment. The reported variation in terms of resistance mechanisms in gliomas and other neoplastic systems reported in primary and recurrent human gliomas underscores this possibility.92-96,98 Cells seen as a subpopulation in the primary tumor become the dominant population in tumors that recur after treatment. Similar results have been obtained by Phillips et al.,77 who found that a subpopulation of cells in a nude mouse xenograft system can expand selectively after treatment with BCNU.
Despite the caveats inherent in analyzing these heterogeneous tumor systems, it is clear that the mechanisms of resistance used by cancer cells to survive treatment with these (and related) compounds are varied. It is equally clear that therapy reduces this heterogeneity, allowing only resistant cells to survive. As our understanding of the biology of gliomas and the body’s response to therapy increases so will the opportunity to design improved therapeutic modalities, which will likely assume many forms. Not only will we need to devise new therapies, but it may be possible to alter the efficacy of current therapies by modulating the expression of genes involved in therapy resistance. Consequently, scientists are looking for novel genes and studying the regulation of DNA repair in response to damage induced by therapy; the regulation of genes that alter the cell cycle and cell growth; and the interplay among cell growth, cell death, and resistance to therapy.
References
- Aebi S, Kurdi-Haidar B, Gordon R, et al: Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res 56:3087-3090, 1996
- Aida T, Cheitlin A, Bodell WJ: Inhibition of O6-alkylguanine-DNA-alkyltransferase activity potentiates cytotoxicity and induction of SCEs in human glioma cells resistant to 1,3-bis(2-chloroethyl)-1-nitrosourea. Carcinogenesis 8:1219-1223, 1987
- Ali-Osman F, Stein DE, Renwick A: Glutathione content and glutathione-S-transferase expression in 1,3-bis(2-chlorethyl)-1-nitrosourea-resistant human malignant astrocytoma cell lines. Cancer Res 50:6976-6980, 1990
- Ban N, Takahashi Y, Takayama T, et al: Transfection of glutathione S-transferase (GST)-š antisense complementary DNA increases the sensitivity of a colon cancer cell line to adriamycin, cisplatin, melphalan, and etoposide. Cancer Res 56:3577-3582, 1996
- Battegay EJ, Raines EW, Seifert RW, et al: TGB- b induces bimodal proliferation of connective tissue cells via complex control of an autocrine PDGF loop. Cell 63:515-524, 1990
- Becker I, Becker K-F, Meyermann R, et al: The multidrug-resistance gene MDR1 is expressed in human glial tumors. Acta Neuropathol Berl 82:516-519, 1991
- Belanich M, Pastor M, Randall T, et al: Retrospective study of a correlation between the DNA repair protein alkyltransferase and survival of brain tumor patients treated with carmustine. Cancer Res 56:783-788, 1996
- Bergelson S, Pinkus R, Daniel V: Intracellular glutathione levels regulate Fos/Jun induction and activation of glutathione S-transferase gene expression. Cancer Res 54:36-40, 1994
- Biaglow JE, Varnes ME, Clark EP, et al: The role of thiols in cellular response to radiation and drugs. Radiat Res 95:437-455, 1983
- Bradford R, Koppel H, Pilkington GJ, et al: Heterogeneity of chemosensitivity in six clonal cell lines derived from a spontaneous murine astrocytoma and its relationship to genotypic and phenotypic characteristics. J Neurooncol 34:247-261, 1997
- Bressler JP, Grotendorst GR, Levitov C, et al: Chemotaxis of rat brain astrocytes to platelet derived growth factor. Brain Res 344:249-254, 1985
- Brown R, Clugston C, Burns P, et al: Increased accumulation of p53 protein in cisplatin-resistant ovarian cell lines. Int J Cancer 55:678-684, 1993
- Brown R, Hirst GL, Gallagher WM, et al: hMLH1 expression and cellular responses of ovarian tumour cells to treatment with cytotoxic anticancer agents. Oncogene 15:45-52, 1997
- Chen ZP, Malapetsa A, Marcantonio D, et al: Correlation of chloroethylnitrosourea resistance with ERCC-2 expression in human tumor cell lines as determined by quantitative competitive polymerase chain reaction. Cancer Res 56:2475-2478, 1996
- Chen ZP, Malapetsa A, McQuillan A, et al: Evidence for nucleotide excision repair as a modifying factor of O6-methylguanine-DNA methyltransferase-mediated innate chloroethylnitrosourea resistance in human tumor cell lines. Mol Pharmacol 52:815-820, 1997
- Christian MC, Pluda JM, Ho PT, et al: Promising new agents under development by the Division of Cancer Treatment, Diagnosis and Centers of the National Cancer Institute. Semin Oncol 24:219-240, 1997
- Clapper ML, Seestaller LM, Tew KD: Induction of glutathione S-transferase alpha RNA in tumor cells following exposure to chlorambucil. Proc Am Assoc Cancer Res 32:361, 1991
- Clark EP: Thiol-induced biochemical modification of chemo- and radioresponses. Int J Radiat Oncol Biol Phys 12:1121-1126, 1986
- Cordon-Cardo C, O’Brien JP, Casals D, et al: Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc Natl Acad Sci USA 86:695-698, 1989
- Dolan ME, Moschel RC, Pegg AE: Depletion of mammalian O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents. Proc Natl Acad Sci USA 87:5368-5372, 1990
- Drummond JT, Anthoney A, Brown R, et al: Cisplatin and adriamycin resistance are associated with MutLalpha and mismatch repair deficiency in an ovarian tumor cell line. J Biol Chem 271:19645-19648, 1996
- Duckett DR, Drummond JT, Murchie AI, et al: Human MutSalpha recognizes damaged DNA base pairs containing O6-methylguanine, O4methylthymine, or the cisplatin-d(GpG) adduct. Proc Natl Acad Sci USA 93:6443-6447, 1996
- Dumenco LL, Warman B, Hatzoglou M, et al: Increase in nitrosourea resistance in mammalian cells by retrovirally mediated gene transfer of bacterial O6-alkylguanine-DNA alkyltransferase. Cancer Res 49:6044-6051, 1989
- el-Deiry WS, Tokino T, Waldman T, et al: Topological control of p21WAF1/CIP1 expression in normal and neoplastic tissues.Cancer Res 55:2910-2919, 1995
- Endicott JA, Ling V: The biochemistry of P-glycoprotein-mediated multidrug resistance. Annu Rev Biochem 58:137-171, 1989
- Fink D, Nebel S, Aebi S, et al: The role of DNA mismatch repair in platinum drug resistance. Cancer Res 56:4881-4886, 1996
- Fink D, Zheng H, Nebel S, et al: In vitro and in vivo resistance to cisplatin in cells that have lost DNA mismatch repair. Cancer Res 57:8141-1845, 1997
- Flaumenhaft R, Abe M, Sato Y, et al: Role of the latent TGF-b binding protein in the activation of latent TGF-b by co-cultures of endothelial and smooth muscle cells. J Cell Biol 120:995-1002, 1993
- Fritsche M, Haessler C, Brandner G: Induction of nuclear accumulation of the tumor-suppressor protein p53 by DNA-damaging agents. Oncogene 8:307-318, 1993
- Frosina G, Rossi O, Arena G, et al: O6-alkylguanine-DNA alkyltransferase activity in human brain tumors. Cancer Lett 55:153-158, 1990
- Fujiwara T, Grimm EA, Mukhopadhyay T, et al: Induction of chemosensitivity in human lung cancer cells in vivo by adenovirus-mediated transfer of the wild-type p53 gene. Cancer Res 54:2287-2291, 1994
- Futscher BW, Erikson LC: Changes in c-myc and c-fos expression in a human tumor cell line following exposure to bifunctional alkylating agents. Cancer Res 50:62-66, 1990
- Gallagher WM, Cairney M, Schott B, et al: Identification of p53 genetic suppressor elements which confer resistance to cisplatin.Oncogene 14:185-193, 1997
- Gerson SL: Modulation of human lymphocyte O6-alkylguanine-DNA alkyltransferase by streptozocin in vivo. Cancer Res 49:3134-3138, 1989
- Gerson SL, Trey JE: Modulation of nitrosourea resistance in myeloid leukemias. Blood 71:1487-1494, 1988
- Gerson SL, Trey JE, Miller K: Potentiation of nitrosourea cytotoxicity in human leukemic cells by inactivation of O6-alkylguanine-DNA alkyltransferase. Cancer Res 48:1521-1527, 1988
- Giaccone G, Gazdar AF, Beck H, et al: Multidrug sensitivity phenotype of human lung cancer cells associated with topoisomerase II expression. Cancer Res 52:1666-1674, 1992
- Gomez-Manzano C, Fueyo J, Kyritsis AP, et al: Characterization of p53 and p21 functional interactions in glioma cells en route to apoptosis. J Natl Cancer Inst 89:1036-1044, 1997
- Hamilton TC, Lai G-M, Rothenberg ML, et al: Mechanisms of resistance to cisplatin and alkylating agents, in Ozols RF (ed): Drug Resistance in Cancer Therapy. Boston: Kluwer Academic Publishers, 1989, pp 151-159
- Hayakawa H, Koike G, Sekiguchi M: Expression and cloning of complementary DNA for a human enzyme that repairs O6-methylguanine in DNA. J Mol Biol 213:739-747, 1990
- Henson JW, Cordon-Cardo C, Posner JB: P-glycoprotein expression in brain tumors. J Neurooncol 14:37-43, 1992
- Huang A, Jin H, Wright JA: Drug resistance and gene amplification potential regulated by transforming growth factor b1 gene expression. Cancer Res 55:1758-1762, 1995
- Hussey AJ, Hayes JD: Characterization of a human class-Theta glutathione S-transferase with activity towards 1-menaphthyl sulfate. Biochem J 286:929-935, 1992
- Jensen PB, Christensen IJ, Sehested M, et al: Differential cytotoxicity of 19 anticancer agents in wild type and etoposide resistant small cell lung cancer cell lines. Br J Cancer 67:311-320, 1993
- Jensen PB, Roed H, Sehested M, et al: Doxorubicin sensitivity pattern in a panel of small-cell lung-cancer cell lines: Correlation to etoposide and vincristine sensitivity and inverse correlation to carmustine sensitivity. Cancer Chemother Pharmacol 31:46-52, 1992
- Kashani-Sabet M, Lu Y, Leong L, et al: Differential oncogene amplification in tumor cells from a patient treated with cisplatin and 5-fluorouracil. Eur J Cancer 26:383-390, 1990
- Kastan MB, Onyekwere O, Sidransky D, et al: Participation of p53 protein in the cellular response to DNA damage. Cancer Res 51:6304-6311, 1991
- Kotoh S, Naito S, Yokomizo A, et al: Enhanced expression of gamma-glutamylcysteine synthetase and glutathione S-transferase genes in cisplatin-resistant bladder cancer cells with multidrug resistance phenotype. J Urol 157:1054-1058, 1997
- Kraker AJ, Moore CW: Elevated DNA polymerase-bactivity in a cis-diamminedichloroplatinum(II) resistant P388 murine leukemia cell line. Cell Lett 38:307-314, 1988
- Kurimoto M, Endo S, Arai K, et al: TM-1 cells from an established human malignant glioma cell line produce PDGF, TGF-a, and TGF-b which cooperatively play a stimulatory role for an autocrine growth promotion. J Neurooncol 22:33-44, 1994
- Lai G-M, Ozols RF, Young RC, et al: Effect of glutathione on DNA repair in cisplatin-resistant human ovarian cancer cell lines. J Natl Cancer Inst 81:535-539, 1989
- Lee SM, Reid H, Elder RH, et al: Inter- and intracellular heterogeneity of O6-alkylguanine-DNA alkyltransferase expression in human brain tumors: Possible significance in nitrosourea therapy. Carcinogenesis 17:637-641, 1996
- Lewis AD, Hayes JD, Wolf CR: Glutathione and glutathione-dependent enzymes in ovarian adenocarcinoma cell lines derived from a patient before and after the onset of drug resistance: Intrinsic differences and cell cycle effects. Carcinogenesis 9:1283-1287, 1988
- Li C, Suardet L, Little JB: Potential role of WAF1/Cip1/p21 as a mediator of TGF-b cytoinhibitory effect. J Biol Chem 270:4971-4974, 1995
- Liang BC: Effects of hypoxia on drug resistance phenotype and genotype in human glioma cell lines. J Neurooncol 29:149-155, 1996
- Lowe SW, Ruley HE, Jacks T, et al: p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 74:957-967, 1993
- Malaise EP, Fertil B, Deschavanne PJ, et al: Initial slope of radiation survival curves is characteristic of the origin of primary and established cultures of human tumor cells and fibroblasts. Radiat Res 111:319-333, 1987
- Marathi UK, Dolan ME, Erickson LC: Extended depletion of O6-methylguanine-DNA methyltransferase activity following O6-benzyl-2´-deoxyguanine or O6-benzylguanine combined with streptozotocin treatment enhances 1,3-bis(2-chloroethyl)-1 nitrosourea cytotoxicity. Cancer Res 54:4371-4375, 1994
- Marathi UK, Dolan ME, Erickson LC: Anti-neoplastic activity of sequenced administration of O6-benzylguanine, streptozotocin, and 1,3-bis(2-chloroethyl)-1 nitrosourea in vitro and in vivo. Biochem Pharmacol 48:2127-2134, 1994
- Marathi UK, Kroes RA, Dolan ME, et al: Prolonged depletion of O6-methylguanine DNA methyltransferase activity following exposure to O6-benzylguanine with or without streptozocin enhances 1,3-bis(2-chloroethyl)-1-nitrosourea sensitivity in vitro. Cancer Res 53:4281-4286, 1993
- Matsumoto T, Tani E, Kaba K, et al: Amplification and expression of a multidrug resistance gene in human glioma cell lines. J Neurosurg 72:96-101, 1990
- Meijer C, Mulder NH, Hospers GAP, et al: The role of glutathione in resistance to cisplatin in a human small cell lung cancer cell line. Br J Cancer 62:72-77, 1990
- Miller AC, Gafner J, Clark EP, et al: Posttranscriptional down-regulation of ras oncogene expression by inhibitors of cellular glutathione. Mol Cell Biol 13:4416-4422, 1993
- Miyazono K, Ichijo H, Heldin C-H: Transforming growth factor-b: Latent forms, binding proteins and receptors. Growth Factors 8:11-22, 1993
- Moses HL, Yang EY, Pietenpol JA: TGF-b stimulation and inhibition of cell proliferation: New mechanistic insights. Cell 63:245-247, 1990
- Mueller H, Eppenberger U: The dual role of mutant p53 protein in chemosensitivity of human cancers. Anticancer Res 16:3845-3848, 1996
- Muller M, Meijer C, Zaman GJR, et al: Overexpression of the gene encoding the multidrug resistance-associated protein results in increased ATP-dependent glutathione S-conjugate transport. Proc Natl Acad Sci USA 91:13033-13037, 1994
- Nabors MW, Griffin CA, Zehnbauer BA, et al: Multidrug resistance gene (MDR1) expression in human brain tumors. J Neurosurg 75:941-946, 1991
- Nehme A, Baskaran R, Aebi S, et al: Differential induction of c-Jun NH2-terminal kinase and c-Abl kinase in DNA mismatch repair-proficient and -deficient cells exposed to cisplatin. Cancer Res 57:3253-3257, 1997
- Norman SA, Rhodes SN, Shapiro JR, et al: Identification of transforming growth factor-b1 binding protein over-expression in BCNU-resistant glioma cells by differential mRNA display. J Neurooncol, submitted
- Nutt CL, Chambers AF, Cairncross JG: Wildtype p53 renders mouse astrocytes resistant to 1,3-bis(2-chloroethyl-)-1nitrosourea despite the absence of a p53-dependent cell cycle arrest. Cancer Res 56:2748-2751, 1996
- Oates TW, Kose KN, Xie JF, et al: Receptor binding of PDGF-AA and PDGF-BB, and the modulation of PDGF receptors by TGF-b, in human periodontal ligament cells. J Cell Physiol 162:359-366, 1995
- Ozols RF, Masuda H, Hamilton TC: Mechanisms of cross-resistance between radiation and antineoplastic drugs. NCI Monogr 6:159-165, 1988
- Pemble S, Schroeder KR, Spencer SR, et al: Human glutathione S-transferase theta (GSTT1): cDNA cloning and the characterization of a genetic polymorphism. Biochem J 300:271-276, 1994
- Perego P, Giarola M, Righetti SC, et al: Association between cisplatin resistance and mutation of p53 gene and reduced baxexpression in ovarian carcinoma cell systems. Cancer Res 56:556-562, 1996
- Phillips PC, Tuma R, Kaufmann SH, et al: Increased expression of O6-alkylguanine-DNA alkyltransferase mRNA in a BCNU-resistant human brain tumor cell line of mer-parentage. Proc Am Assoc Cancer Res 32:355, 1991
- Phillips WP, Jr., Willson JKV, Markowitz SD, et al: O6-methylguanine-DNA methyltransferase (MGMT) transfectants of a 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU)-sensitive colon cancer cell line selectively repopulate heterogeneous MGMT+/MGMT-xenografts after BCNU and O6-benzylguanine plus BCNU. Cancer Res 57:4817-4823, 1997
- Pieper RO, Futscher BW, Dong Q, et al: Effects of streptozotocin/bis-chloroethylnitrosourea combination therapy on O6-methylguanine DNA methyltransferase activity and mRNA levels in HT-29 cells in vitro. Cancer Res 51:1581-1585, 1991
- Potapova O, Haghighi A, Bost F, et al: The Jun kinase/stress-activated protein kinase pathway functions to regulate DNA repair and inhibition of the pathway sensitizes tumor cells to cisplatin. J Biol Chem 272:14041-14044, 1997
- Price BD, Park SJ: DNA damage increases the levels of MDM2 messenger RNA in wtp53 human cells. Cancer Res 54:896-899, 1994
- Puchalski RB, Fahl WE: Expression of recombinant glutathione S-transferase š, Ya, or Yb1 confers resistance to alkylating agents.Proc Natl Acad Sci USA 87:2443-2447, 1990
- Roth JA, Cristiano RJ: Gene therapy for cancer: What have we done and where are we going? J Natl Cancer Inst 89:21-39, 1997
- Rydberg B, Spurr N, Karran P: cDNA cloning and chromosomal assignment of the human O6-methylguanine-DNA methyltransferase. cDNA expression in Escherichia coli and gene expression in human cells. J Biol Chem 265:9563-9569, 1990
- Scanlon KJ, Kashani-Sabet M: Elevated expression of thymidylate synthase cycle genes in cisplatin-resistant human ovarian carcinoma A2780 cells. Proc Natl Acad Sci USA 85:650-653, 1988
- Scanlon KJ, Wang WZ, Han H: Cyclosporin A suppresses cisplatin-induced oncogene expression in human cancer cells. Cancer Treat Rev 17 (suppl A):27-35, 1990
- Scheck AC, Beikman MK, Korn MC, et al: Regional analysis of genes potentially involved in resistance to BCNU in human malignant gliomas. Proc Am Assoc Cancer Res 32:358, 1991
- Scheck AC, Mehta BM, Beikman MK, et al: BCNU-resistant human glioma cells with overrepresentation of chromosomes 7 and 22 demonstrate increased copy number and expression of platelet-derived growth factor genes. Genes Chromosomes Cancer 8:137-148, 1993
- Scheck AC, Mihara Y, Beikman MK, et al: Regional analysis of human malignant gliomas: Expression of genes mapped to chromosomes 7 and 22. Neurology 41:158, 1991
- Scheck AC, Mihara Y, Shapiro JR: Regional heterogeneity in human malignant gliomas occurs in anaplastic astrocytomas as well as glioblastomas multiforme. Proc Am Assoc Cancer Res 31:306, 1990
- Scheck AC, Norman SA, Shapiro JR: Identification of genes involved in BCNU resistance in human malignant gliomas using differential mRNA display. Proc Am Assoc Cancer Res 36:326, 1995
- Shapiro JR: Cellular characterization and BCNU resistance of freshly resected and early passage human glioma cells. Prog Neuropathol 6:133-143, 1986
- Shapiro JR: Biology of gliomas: Heterogeneity, oncogenes, growth factors. Semin Oncol 13:4-15, 1986
- Shapiro JR, Ebrahim SAD, Mohamed AN, et al: BCNU-sensitivity in parental cells and clones from four freshly resected near-diploid human gliomas: An astrocytoma, an anaplastic astrocytoma and two glioblastomas multiforme. J Neurooncol 15:209-227, 1993
- Shapiro JR, Mehta BM, Ebrahim SAD, et al: Tumor heterogeneity and intrinsically chemoresistant subpopulations in freshly resected human malignant gliomas, in Sudilovsky O, Pitot HC, Liotta LA (eds): Boundaries Between Promotion and Progression During Carcinogenesis. New York: Plenum Press, 1991, pp 243-262
- Shapiro JR, Mehta BM, Fiola MR, et al: Intrinsically chemo- and radio-resistant subpopulations identified in freshly resected human gliomas become the dominant population in recurrent tumor samples. Neurology 40 (suppl 1):395, 1990
- Shapiro JR, Pu P-Y, Mohamed AN, et al: Chromosome number and carmustine sensitivity in human gliomas. Cancer 71:4007-4021, 1993
- Shapiro JR, Pu P-Y, Shapiro WR: Resistant cell types in human gliomas, in Salmon S, Trent JM (eds): Tumor Cell Cloning. Orlando: Grune & Stratton, 1984, pp 133-142
- Shapiro JR, Shapiro WR: The subpopulations and isolated cell types of freshly resected high grade human gliomas: Their influence on the tumor’s evolution in vivo and behavior and therapy in vitro. Cancer Metastasis Rev 4:107-124, 1985
- Shapiro JR, Shapiro WR: BCNU resistance cosegregates with PDGF autocrine growth in primary and recurrent tumor cells carrying a specific chromosome complement. J Neurooncol 5:180, 1987
- Shapiro JR, Yung W-KA, Shapiro WR: Isolation, karyotype, and clonal growth of heterogeneous subpopulations of human malignant gliomas. Cancer Res 41:2349-2359, 1981
- Shea TC, Clafin G, Comstock KE, et al: Glutathione transferase activity and isoenzyme composition in primary human breast cancers. Cancer Res 50:6848-6853, 1990
- Silber JR, Blank A, Bobola MS, et al: Lack of the DNA repair protein O6-methylguanine-DNA methyltransferase in histologically normal brain adjacent to primary human brain tumors. Proc Natl Acad Sci USA 93:6941-6946, 1996
- Silber JR, Mueller BA, Ewers TG, et al: Comparison of O6-methylguanine-DNA methyltransferase activity in brain tumors and adjacent normal brain. Cancer Res 53:3416-3420, 1993
- Smith ML, Kontny HU, Zhan QM, et al: Antisense GADD45 expression results in decreased DNA repair and sensitizes cells to u.v.-irradiation or cisplatin. Oncogene 13:2255-2263, 1996
- Smith MT, Evans CG, Doan-Setzer P, et al: Denitrosation of 1,3-bis(2-chloroethyl)-1-nitrosourea by class mu glutathione transferases and its role in cellular resistance in rat brain tumor cells. Cancer Res 49:2621-2625, 1989
- Sriram R, Ali-Osman F: Enzymes of glutathione biosynthesis and metabolism in human malignant astrocytomas. Proc Am Assoc Cancer Res 32:428, 1991
- Taipale J, Saharinen J, Hedman K, et al: Latent transforming growth factor-b and its binding protein are components of extracellular matrix microfibrils. J Histochem Cytochem 44:875-889, 1996
- Tano K, Shiota S, Collier J, et al: Isolation and structural characterization of a cDNA clone encoding the human DNA repair protein for O6-alkylguanine. Proc Natl Acad Sci USA 87:686-690, 1990
- Tsuchida S, Sekine Y, Shineha R, et al: Elevation of the placental glutathione S-transferase form (GST-š) in tumor tissues and the levels in sera of patients with cancer. Cancer Res 49:5225-5229, 1989
- Van Laethem JL, Resibois A, Adler M, et al: Localization of transforming growth factor-b1 precursor and latent TGF-b1 binding protein in colorectal adenomas. Dig Dis Sci 41:1741-1748, 1996
- von Wronski MA, Shiota S, Tano K, et al: Structural and immunological comparison of indigenous human O6-methylguanine-DNA methyltransferase with that encoded by a cloned cDNA. J Biol Chem 266:1064-1070, 1991
- Waldman T, Kinzler KW, Vogelstein B: p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res 55:5187-5190, 1995
- Waldman T, Zhang Y, Dillehay L, et al: Cell-cycle arrest versus cell death in cancer therapy. Nature Medicine 3:1034-1036, 1997
- Wang YY, Teicher BA, Shea TC, et al: Cross-resistance and glutathione-S-transferase-š levels among four human melanoma cell lines selected for alkylating agent resistance. Cancer Res 49:6185-6192, 1989
- Wasenius VM, Jekunen A, Monni O, et al: Comparative genomic hybridization analysis of chromosomal changes occurring during development of acquired resistance to cisplatin in human ovarian carcinoma cells. Genes Chromosomes Cancer 18:286-291, 1997
- Yamakage A, Kikuchi K, Smith EA, et al: Selective upregulation of platelet-derived growth factor a receptors by transforming growth factor b in scleroderma fibroblasts. J Exp Med 175:1227-1234, 1992
- Yung W-K, Shapiro JR, Shapiro WR: Heterogeneous chemosensitivities of subpopulations of human glioma cells in culture.Cancer Res 42:992-998, 1982
- Zaman GJR, Flens MJ, van Leusden MR, et al: The human multidrug resistance-associated protein MRP is a plasma membrane drug-efflux pump. Proc Natl Acad Sci USA 91:8822-8826, 1994